

1 Introduction
Interest in transuranic coordination chemistry is driven not only by scientific curiosity to understand the fundamentals of all of the accessible elements in the Periodic Table, but also through a recognition that a comprehensive grasp of the basic electronic structure, bonding, and reactivity of the early to middle actinides (Th-Cm) is vital to support the development of advanced nuclear fuel cycles, and novel separation and waste remediation processes that are crucial to allow nuclear power to continue to play a significant role in meeting global energy demands [1].
Although investigations on aqueous and non-aqueous uranium chemistry remain dominant [2], research on transuranium chemistry is witnessing some great developments, but our current understanding of the electronic structure and bonding interaction of these elements is from aqueous systems for which the choice of ligands are limited to water-stable “hard” donors that will displace the water/hydroxy ligands and coordinate to the metal centre [3]. However, despite the synthesis of promising precursors [4], anhydrous chemistry of Np and Pu is still in the early stages of development.
Phosphine oxides have been used in the nuclear industry as extraction agents for waste reprocessing (e.g., PUREX process). Hence, academic research into the coordination chemistry of f-elements with PO donor ligands has been particularly intense. Many research groups have turned their attention to the bidentate bis(diphenylphosphino)methane dioxide (DPPMO2) ligand and its coordination with uranium [5], but no transuranic complexes have been reported so far. Previously, we have shown that the weakly coordinating oxoanions [ReO4]− and [TcO4]− (as well as Cl−) will fill a fifth coordination site yielding complexes of general formula [UO2(DPPMO2)2X][X] (where X = [ReO4]−, [TcO4]− and Cl−), with the X− anion acting in both monodentate coordinated and uncoordinated modes [5b,f]. As part as our investigations into transuranic phosphine oxide complexes, we prepared a neptunyl complex with the DPPMO2 ligand as [NpO2(DPPMO2)2Cl]2[NpO2Cl4] (1).
Further investigations to test the reactivity and stability of our recently prepared anhydrous precursor [NpO2Cl2(thf)]n under different experimental conditions have been carried out to compare with the reactivity of [UO2Cl2(thf)2]2. Unlike the synthesis of [UO2Cl2(thf)2]2, the preparation of [NpO2Cl2(thf)]n remains challenging because of the rapid reduction of Np(VI) to Np(V) in organic solvents. Solid-state characterisation of both compounds showed differences between both complexes with the neptunyl compound crystallising as a polymer compared to a dimer for the uranyl species [4b,4c]. Our group has extensively studied the reactivity of [UO2Cl2(thf)2]2 with phosphorus containing ligands R3PX (RCy, Ph and XO, NH) [6] so we were particularly interested in comparing the reactivity of phosphinimine ligands with [NpO2Cl2(thf)]n. Reaction of [NpO2Cl2(thf)]n with Ph3PNH led to the characterisation of [Ph3PNH]2[NpO2Cl4] (2).
Both crystals structures of 1 and 2 indicated that the anion [NpO2Cl4]2− seems to be unusually stable in aprotic solvents. We comment here on the relative the stability of [AnO2Cl4]2− along the actinides series and identify the conditions in which this anion is chemically stable.
2 Results and discussions
2.1 [NpO2(DPPMO2)2Cl]2[NpO2Cl4]
A MeOH solution of DPPMO2 (2 eq.) was added to a solution of Np(VI) in HCl yielding a pale yellow solution. Crystals of [NpO2(DPPMO2)2Cl]2[NpO2Cl4] (Fig. 1) were obtained by slow evaporation of the solvent at room temperature. To our knowledge, this complex is the first neptunium compound containing another Np(VI) counterion. The structure of 1 revealed a neptunium(VI) mixed salt comprising a dianionic complex [NpO2Cl4]2− (1a) and two monocationic pentagonal bipyramidal [NpO2(DPPMO2)2Cl]+ (1b) (Fig. 1). Selected bond distances and angles are given in Table 1.
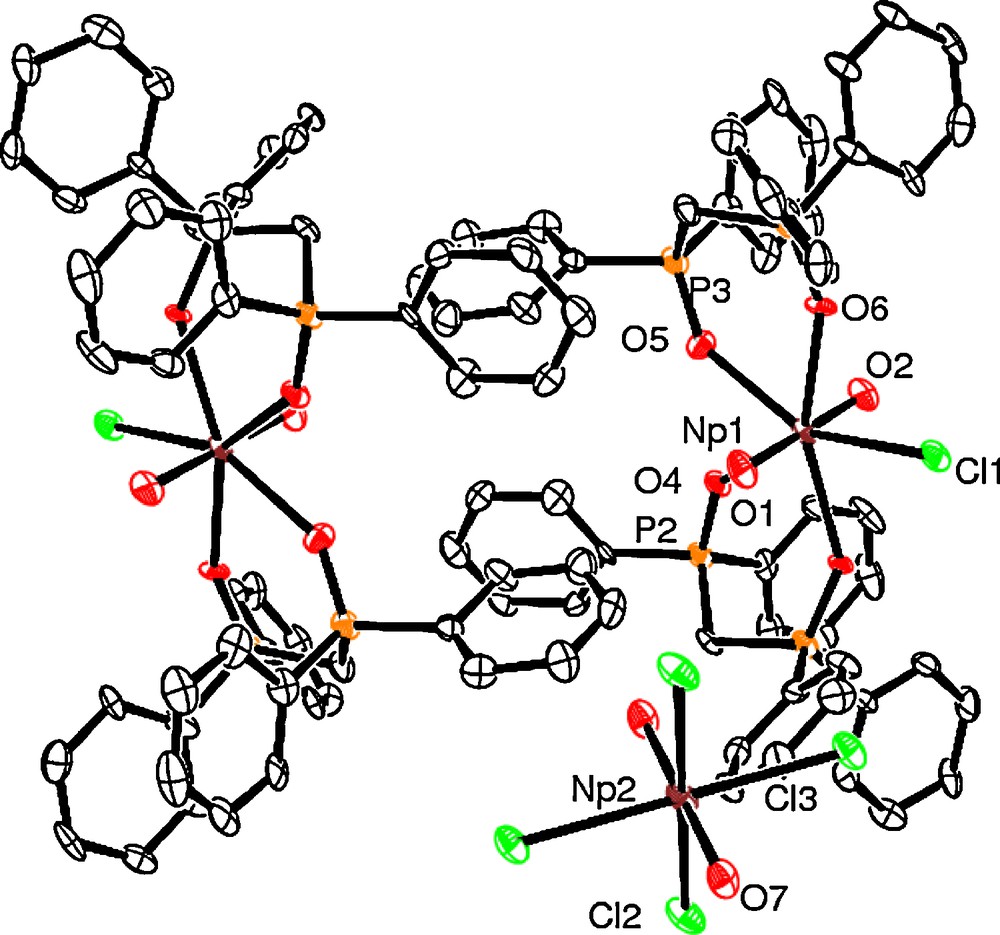
ORTEP representation of [NpO2(DPPMO2)2Cl]2[NpO2Cl4] (1).
Selected bond lengths [Å] and angles [°] for 1.
| Bond length (Å) | Bond angle (°) | ||
| 1b | |||
| Np(1)-O(1) | 1.766(8) | O(1)-Np(1)-O(2) | 179.4(4) |
| Np(1)-O(2) | 1.773(8) | ||
| Np(1)-Cl(1) | 2.670(3) | ||
| Np(1)-O(3) | 2.388(7) | ||
| Np(1)-O(4) | 2.415(7) | ||
| Np(1)-O(5) | 2.373(8) | ||
| Np(1)-O(6) | 2.381(7) | ||
| 1a | |||
| Np(2)-O(7) | 1.751(7) | O(7)-Np(2)-O(7) | 180.0(5) |
| Np(2)-Cl(2) | 2.652(3) | Cl(3)-Np(2)-Cl(3) | 180 |
| Np(2)-Cl(3) | 2.666(3) |
The Np atom in 1a is located on an inversion centre, with half the molecule contained in the unit cell, exhibiting identical NpO distances (1.751(7) Å), which are within the range expected for Np–Oyl bond lengths [7]. Np–Cl distances are different with Np(2)–Cl(2) (2.652(3) Å) being slightly shorter than Np(2)–Cl(3) (2.666(3) Å). The structure of neptunyl tetrachloride complex 1a has previously been reported in the mixed valence species Cs7[NpVO2][NpVIO2]2Cl12, [8], Cs2[NpO2Cl4] [9] and [NBu4]2[NpO2Cl4] [10]. Comparison of the bond lengths in 1a with those found in Cs7[NpVO2][NpVIO2]2Cl12 is precluded due to the extensive disorder present in the structure in the mixed valence compound. In contrast with 1a, in [NBu4]2[NpO2Cl4], the NpO bonds are inequivalent by symmetry but the Np–O distances (1.733(5) and 1.740(6) Å) are indistinguishable and the ONpO angle is close to linearity (178.6(3)°) as in 1a (180°).
To the best of our knowledge, [NpO2(DPPMO2)2Cl]+ (1b) is the first seven-coordinated neptunium(VI) complex reported so far. In 1b, the Np–Oyl distances (average 1.769(8) Å) are statistically similar to the one found in 1a and other structurally characterised neptunyl complexes (1.767(10) Å for [NpO2Cl2(thf)]n [4b], 1.739(10) Å and 1.751(18) Å for [NpO2Cl2(TPPO)(NO3)] and [NpO2Cl2(TPPO)2] [11] respectively). AnO distances are statistically the same as the one observed in seven coordinated uranyl-DPPMO complexes that have UO bond distances of average 1.7555(12) Å [5a,5e]. The ONpO unit is linear (179.4(4)°). The NpCl bond distance of 2.670(3) Å is similar to the ones in 1a, but is longer than the Np–Cl distances found in [NpO2Cl2(TPPO)2] (average of 2.6335(3) Å). As previously observed in the [UO2(DPPMO2)2X]+ cation, the DPPMO2 groups are not symmetrical, displaying Np–OP=O bond lengths in the range between 2.373(8) to 2.415(7) Å. The average Np–O(P=O) distance (2.389(7) Å) exhibited by 1b is longer than that in the Np(VI) tetragonal bipyramidal complex in [NpO2(TPPO)4][ClO4]2 (average of 2.284(6) Å) [11] and is longer than the average Np–O(P=O) bond length found in trans-[NpO2Cl2(TPPO)2] (2.2745(24) Å) [7a]. One of the DPPMO2 ligands in 1b has Np–O(P=O) bond lengths that are statistically shorter (Np(1)–O(5), 2.373(8) Å; Np(1)–O(6) 2.381(7) Å) than the longest Np–O(P=O) distance found in the second chelating DPPMO2 group (Np(1)–O(4), 2.415(7) Å), whilst the P–O bond lengths are in the range 1.479(8)–1.506(8) Å (Table 2).
Selected bond lengths [Å] and angles [°] for 2.
| Bond lengths (Å) | Bond angles (°) | ||
| Np(1)-O(1) | 1.752(3) | O(1)-Np-O(2) | 179.60(14) |
| Np(1)-O(2) | 1.748(3) | Cl(1)-Np-Cl(3) | 176.49(3) |
| Np(1)-Cl(1) | 2.6469(11) | Cl(2)-Np-Cl(4) | 179.70(3) |
| Np(1)-Cl(3) | 2.6521(10) | ||
| Np(1)-Cl(2) | 2.6621(11) | ||
| Np(1)-Cl(4) | 2.6640(11) | ||
| P(1)-N(1) | 1.628(4) | ||
| N(1)-H(1) | 0.859(19) | ||
| N(1)-H(2) | 0.853(19) |
Perhaps of most interest in the structural characterisation of 1 is a comparison with the uranyl analogue, [UO2(DPPMO2)2Cl]Cl [5b], in order to assess the impact that the identity of the metal ion (U, Np) has upon the structure of the [AnO2(DPPMO2)2Cl]+ (AnU, Np) complex cation. Of course, this must be approached with caution because the identity of the non-coordinating anion in 1 ([NpO2Cl4]2−) is different to that in [UO2(DPPMO2)2Cl]+ (Cl−). Crystal packing forces and unit cell dimensions/space group differences because of the different anion may well have an effect on bond lengths and angles in the first coordination sphere of the metal ion. The U–O(P=O) bond distance (average of 2.39(2) Å [5b], in [UO2(DPPMO2)2Cl]+ is statistically the same as the Np–O(P=O) distance (average of 2.389(7) Å) in 1b). The average bite angle of the DPPMO2 groups in 1b (70.5(4) Å) and [UO2(DPPMO2)2Cl]+ (70.5(2) Å) [5b] are identical, despite the smaller size of neptunium in comparison to uranium. A comparison of the bond lengths in the OAnO unit in 1b (AnNp) (1.766(8) and 1.773(8) Å) and [UO2(DPPMO2)2Cl]+ (1.762(7) Å) [5b], reveals that the neptunyl(VI) bond distances are statistically the same as the uranyl(VI) bond length. It is intriguing that this structural similarity is present, given the f electron located in the {NpO2}2+ moiety and the smaller size of neptunium. However, the Np–Cl bond distance (2.670(3) Å) in 1b is shorter than the U–Cl bond length (2.710(2) Å) [5b] in [UO2(Cl)(DPPMO2)2]+. Neglecting non-bonding ligand–ligand interactions, this difference may in part be explained by the “actinide contraction,” where the chloride ion is drawn closer towards the metal ion as it becomes smaller in passing from U to Np.
The room temperature 31P{1H} NMR spectrum of 1 in CD2Cl2:CD3OD is shown in Fig. 2 and displayed a broad signal at −1.5 ppm. Free DPPMO2 ligand resonates at 25 ppm whilst the bound DPPMO2 ligand in the f0 system [UO2(DPPMO2)2Cl]Cl exhibits a broad singlet at 43.56 ppm at room temperature [5b]. Both the lineshape and the shift to negative field provide evidence that the DPPMO2 molecule is bonded to the f1 {NpO2}2+ ion in solution. Chemical exchange of the [X]− anion in the [UO2(DPPMO2)2X]X (XBr, Cl) compounds is well known and it is highly likely that the bound chloride ion in 1b is also in chemical exchange in solution on the NMR timescale. It is unclear whether the broad lineshape of the NMR signal of 1 is caused by this phenomenon or by a combination of chemical exchange and/or the line broadening effect an NMR nucleus undergoes when interacting with a paramagnetic metal ion. The possible fluxionality of 1b in solution was not probed further using low temperature NMR spectroscopy. It was anticipated that the known temperature dependence of isotropic shifts [12] may further complicate the assignment of the peaks in the spectra.
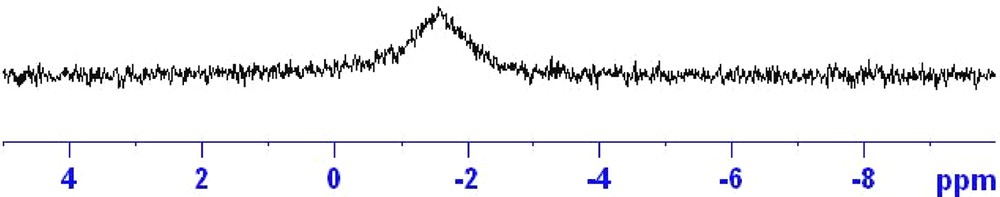
31P{1H} NMR of crystals of 1 in CD2Cl2:CD3OD (50:50).
The electronic absorption spectrum crystals of 1 dissolved in CD2Cl2/CD3OD was recorded and is shown Fig. 3. The UV/vis/nIR spectrum showed the presence of three different oxidation states of neptunium, Np(IV) (bands between 680 and 980 nm), Np(V) (sharp band at 980 nm) and Np(VI) (broad band between 1170 and 1270 nm). Np(VI) readily reduces into Np(IV) and Np(V). In order to understand the behaviour of Np(VI) in organic solvents, absorption spectra of a {NpO22+} stock (NpO2Cl2.xH2O) solution were recorded in a MeOH or THF solution (Fig. 4a and 4b). As expected, dissolution of the “NpO2Cl2xH2O” residue in the reducing solvent MeOH produces Np(V), as evidenced by the characteristic sharp absorption band at 980 nm (Fig. 4a).

UV-vis-nIR spectrum of crystals of 1 in CD2Cl2:CD3OD (50:50).
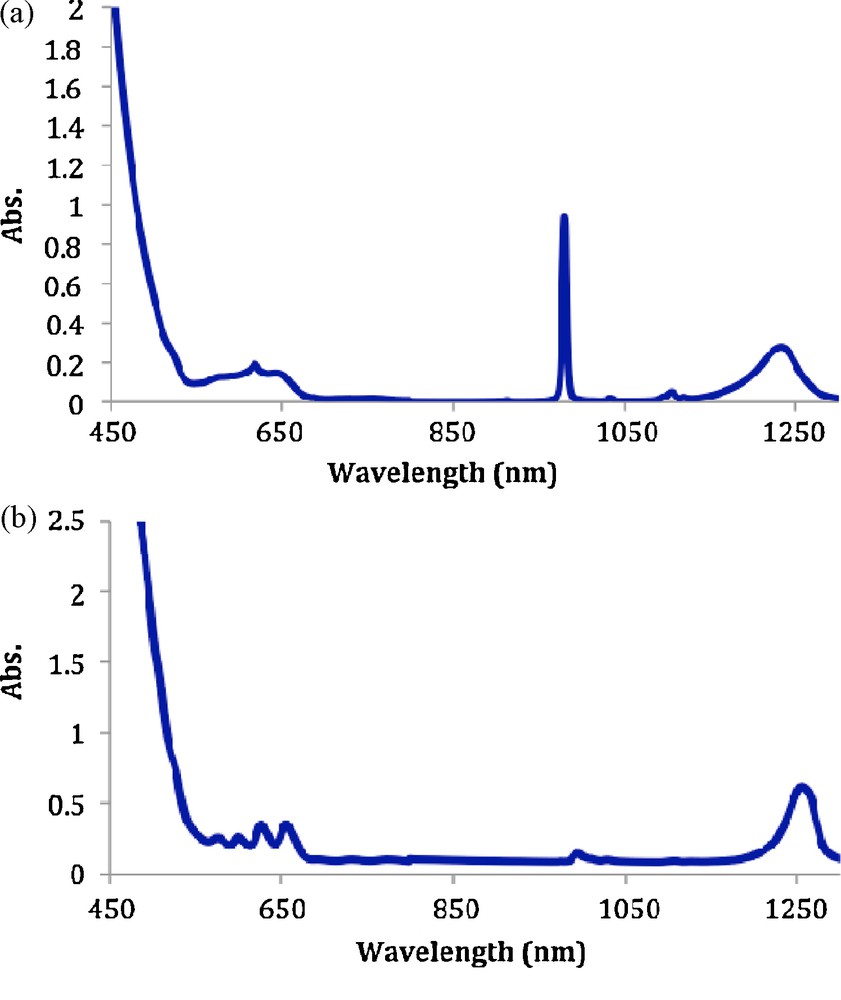
a: UV-vis-nIR spectrum of NpO2Cl2.xH2O in MeOH. b: UV-vis-nIR spectrum of NpO2Cl2.xH2O in THF.
Crystalline 1 is relatively stable with respect to reduction in THF but is reduced in MeOH. Therefore, this reduction occurs prior to any complexation reaction (for example, with DPPMO2) if the reducing alcohol is required to dissolve the ligand molecule. The presence of Np(VI) in the spectrum of 1 is confirmed by the two broad bands in the nIR region between 1170 and 1270 nm corresponding to the two Np(VI) chromophores in 1 (Fig. 3).
2.2 [Ph3PNH2]2[NpO2Cl4](2)
Ph3PNH was added to a THF-d8 solution of [NpO2Cl2(thf)]n which had been prepared from dry [NpO2(OH)2] treated with HCl/Et2O and SiMe3Cl (Scheme 1). Small crystals were formed after a few days from a CH2Cl2 solution. The structure of [Ph3PNH2]2[NpO2Cl4] is shown Fig. 5. Selected bond distances and angles are displayed in Table 2. In contrast to 1a, [NpO2Cl4]2− in 2a does not have a crystallographically imposed inversion centre. The NpO distances are 1.748(3) and 1.752(3) Å and are within the range expected for Np-Oyl complexes. Np–Cl (average 2.6600(11) Å) bond lengths are comparable to those reported for 1a. There are two crystallographically distinct [Ph3PNH2]+ cations with the PN bond distances (1.628(4) and 1.614(4) Å) comparable to the values obtained in [Ph3PNH2]2[OsCl6].2CH3CN [13a], [Ph3NH2]Cl.CH3CN, [13b] [Ph3PNH2]Br.CH2Cl2 [13c], [Ph3PNH2][SCN] [13d]. The phosphonium cations interact with the [NpO2Cl4]2− anion through the NH…Cl hydrogen bonding interactions (Fig. 6).
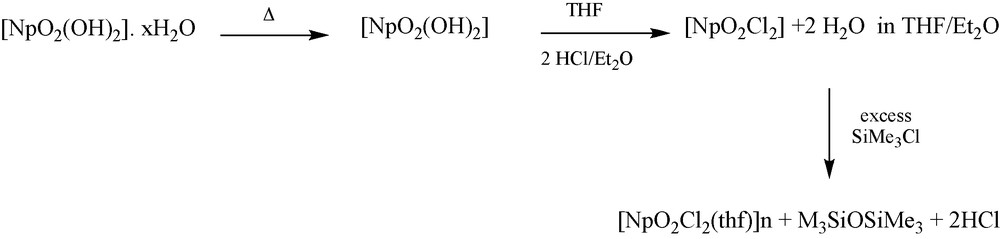
Preparation of a solution of [NpO2Cl2(thf)]n.
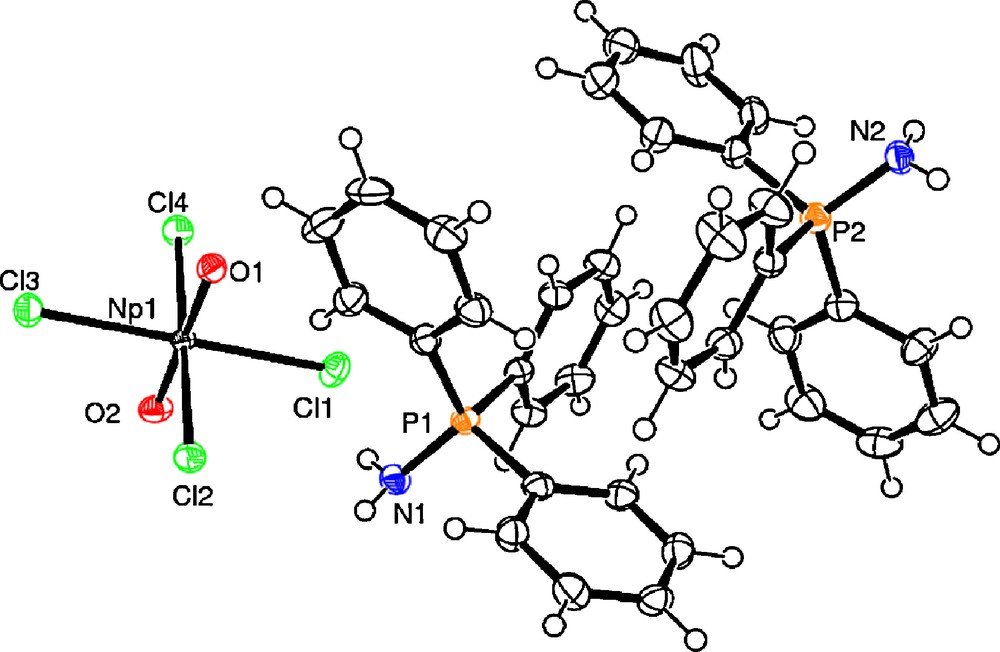
ORTEP representation of [Ph3PNH2]2[NpO2Cl4] (2).
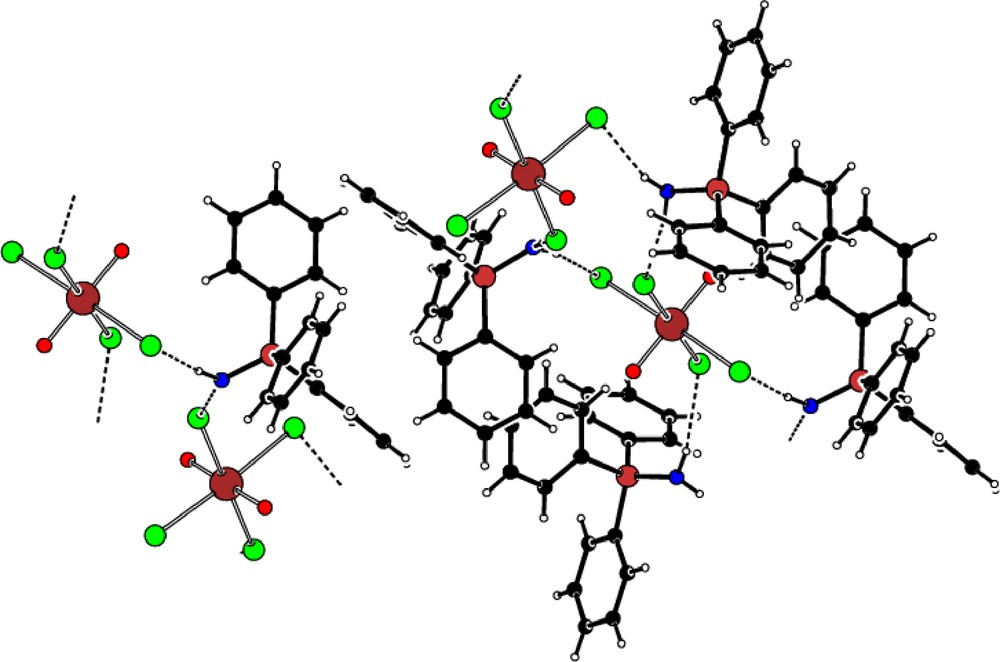
View of [Ph3PNH2]2[NpO2Cl4] showing the hydrogen bonding between the N-H protons of the [Ph3PNH2]+ cations and the Cl atoms on [NpO2Cl4]2−.
UV/Vis/nIR spectra of 2 in THF (Fig. 7) exhibit two broad bands at 1193 and 1219 nm characteristic of Np(VI) complexes and may indicate the presence of two Np(VI) compounds in solution. The 31P{1H} NMR spectrum of the crystals dissolved in CD2Cl2 exhibited a singlet at 36.1 ppm corresponding to the [Ph3NH2]+ salt.
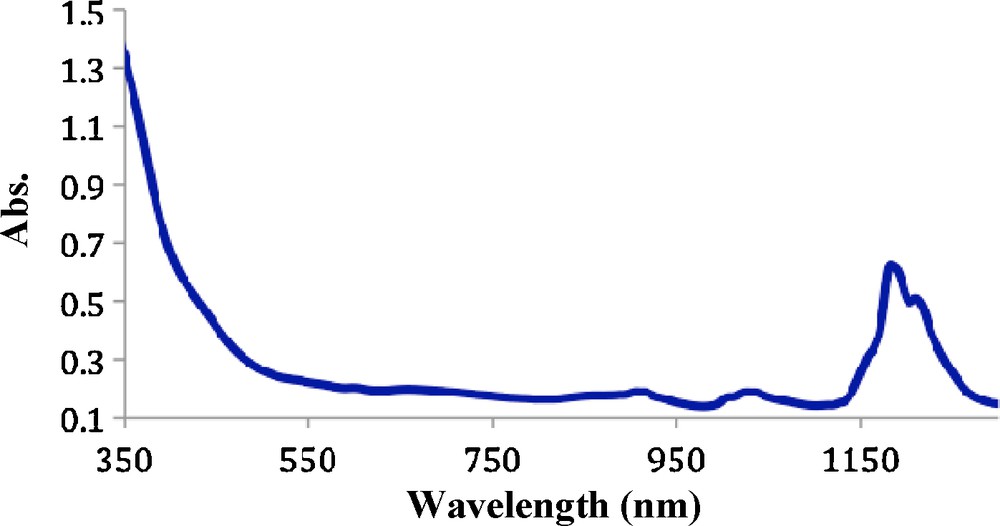
UV-vis-nIR spectrum of 2 in THF.
Interestingly, reaction of [PuO2Cl2(thf)2]2 crystals with Ph3PNH in THF at room temperature yielded [Ph3PNH2]2[PuO2Cl4] after recrystallisation in CH2Cl2 [14]. These results indicate the stability of [AnO2Cl4]2− compared to [AnO2Cl2(thf)2] for Np or Pu in contrast to the analogous uranium complex, which yielded the [UO2Cl2(Ph3PNH)2] complex under air-, moisture- and HCl free conditions [6].
Syntheses of [NpO2Cl4]2− and [PuO2Cl4]2− have been previously reported by reacting a stock solution of {AnO2}2+ in HCl with tetrabutylammonium chloride or caesium chloride in HCl solution, respectively [9,10,15]. However, for compound 1, the only source of chloride available in solution comes from the stock solution of NpO2Cl2.xH2O in conc. HCl. Addition of the DPPMO2 ligand in an aprotic solvent yields the complex [NpO2(DPPMO2)2Cl]2+ liberating Cl− to form the stable [NpO2Cl4]2− counteranion. This would explain the presence of [NpO2Cl4]2− in the solution in the case of 1. Further investigations will be carried out to prepare [NpO2(DPPMO2)2Cl][Cl] by dissolving [NpO2(OH)2].xH2O in a dilute HCl solution (1–2 M). Dissolution of [NpO2(OH)2].xH2O in conc. HCl might directly form [NpO2Cl4]2−. Recent reports on uranyl compounds [UO2(H2O)5]2+ showed that when the concentration of HNO3 is increased from 9 to 14.5 M then the water molecules are displaced with NO32− to go from [UO2(H2O)5]2+ in 1 M HClO4 to [UO2(H2O)(NO3)5]− (9 M HNO3) and [UO2(NO3)3]− (14.5 M) [16].
In the case of the synthesis of compound 2, if moisture was present, coordinated THF in [NpO2Cl2(thf)]n would exchange readily with water and Ph3NH would be converted to Ph3PO and NH3. However, there was no evidence for the presence of Ph3PO in the 31P{1H} NMR spectrum. It has been observed that, in the presence of chloride ions, the phosphinimine ligand can form [Ph3NH2]+Cl− [17], providing a source of chloride for the {NpO2}2+ cation to form [NpO2Cl4]2−. No Ph3PNH was observed in the 31P{1H} NMR spectrum, suggesting that the whole of the ligand has been converted to the [Ph3NH2+]Cl−, the chloride ion probably coming from SiMe3Cl (Scheme 1) still present in the neptunium starting material solution, which then dissociates to form [NpO2Cl4]2−.
3 Conclusion
In summary, we have prepared and characterized two neptunyl complexes containing the [NpO2Cl4]2− moiety as a counteranion.
Only a few examples of the analogous mixed cations-anions uranium compounds containing the [UO2Cl4]2− [18] anion have been reported presumably because, as a Lewis acid, it usually readily reacts with additional organic ligands to form single uranyl complexes [19,20]. In most cases, when [UO2Cl4]2− has been isolated it has been in reactions with soft donor ligands where the coordination does not occur.
The solid state comparison of the [AnO2Cl4]2− anions does not show any evidence of the actinyl contraction since the bond length and angle along the actinides series are statistically the same (Table 3). However, we believe that the solution chemistry differs from one another since uranium reacts easily with both ligands studied in this report to form uranyl complexes.
Comparison of bond lengths [Å] and angles [°] in [AnO2Cl4]2− ions.
| [UO2Cl4]2− | [NpO2Cl4]2− | [PuO2Cl4]2− | |
| [UO2Cl4]3[UIVCl(dmf)7]2 | Cs2NpO2Cl4 | [Ph3PNH]2[PuO2Cl4] | |
| An-Oyl (Å) | 1.77(1) | 1.775(17) | 1.752(3) |
| An-Cl (Å) | 2.669(5)– 2.672(5) | 2.653(3) | 2.6646(8) |
| OAnO (°) | 180 | 180.0(8) | 180.0(3) |
| Ionic radii (Å) | {UO2}2+ 0.83 | {NpO2}2+ 0.82 | {PuO2}2+ 0.81 |
| Reference | 20 | 9 | 14 |
Complex 1 exhibited a shorter Np–Cl bond length compared to the uranium compound and the production of [AnO2Cl4]2−, where An = Np or Pu, seems to be facilitated. This could be explained by a softening of the metal along the transuranium series.
4 Experimental
4.1 General experimental
Caution! 237Np is a high specific activity radionuclide and all manipulations with this radioisotope should be undertaken in a properly regulated and controlled radiochemical laboratory.
Experiments were carried out either in a radiological fume hood, under an argon atmosphere using standard Schlenk line techniques and apparatus when necessary, or in an inert atmosphere argon glovebox. All solvents employed were reagent grade and dried by refluxing over appropriate drying agents.
4.2 Instrumentation
31P{1H} NMR spectra were recorded on a Bruker Avance Ultrashield 400 instrument at 161.91 MHz. UV/vis/nIR spectra were measured on a Varian Cary 500.
Single crystal X-ray diffraction data of 1 were obtained on a Bruker AXS SMART diffractometer (MoKα radiation, λ = 0.71073 Å) at 100(2) K. Data collection and structure refinements were achieved using standard Bruker AXS control and integration software and SHELXTL for all compounds.
Diffraction data for 2 were collected at 100 K using synchrotron radiation at Daresbury SRS, UK (Station 9.8) with a Bruker-Nonius APEX11 CCD diffractometer (λ = 0.6934 Å). Crystals of 2 were surrounded by oil (Fomblin, perfluoro polyether) and mounted inside a polyimide capillaries coated by varnish providing triple containment. The structure was solved by direct methods using SIR92 and refined using SHELXL97 [21,22]. All non-hydrogen atoms not exhibiting disorder were refined anisotropically, while hydrogen atoms were included in calculated positions. All presented ORTEP plots show probability ellipsoids of 50% [23].
4.3 Syntheses
4.3.1 Synthesis of [NpO2Cl2] in conc. HCl
2.4 mL of an HNO3 stock solution of {NpVO2}+ (50 mg, 0.21 mmol of 237Np purchased from CERCA LEA- France-) was evaporated to dryness under a heat lamp. An aqueous {NpVIO2}2+ solution was generated by dissolving the Np(V) solid in conc. HClO4 (70%, 5 ml) and heating until fuming. Dilution of this yellow acidic solution with 0.5 mL H2O yielded a pink solution. UV/vis/nIR spectroscopy was used to confirm the presence of {NpVIO2}2+ (major f–f transition at 1223 nm) and the absence of {NpVO2}+ (major f–f transition at 980 nm). Brown solid neptunyl hydroxide, [NpO2(OH)2].xH2O, was precipitated from solution through the addition of NaOH (6 M). This brown precipitate of [NpO2(OH)2].xH2O was washed twice with H2O, and dissolved in 37% HCl (< 1 ml).
4.3.2 Synthesis of [NpO2Cl(DPPMO)2]2[NpO2Cl4] (1)
A MeOH solution of DPPMO2 (22 mg, 0.052 mmol) was added to an aliquot of the stock solution of NpO2Cl2 in 37% HCl (6.32 mg, 0.026 mmol of 237Np). A white precipitate formed immediately and MeOH was added until the precipitate dissolved yielding a pale yellow solution. The vial containing the solution was left to stand at room temperature for 5 days. Slow evaporation of the solvent afforded yellow crystals of 1. 31P{1H} NMR (CD2Cl2:CD3OD): δ −1.5 ppm (broad s, w1/2 93.55 Hz)
4.3.3 Synthesis of [Ph3PNH2]2[NpO2Cl4] (2)
The synthesis of [NpO2(OH)2].xH2O has been described above. The brown precipitate of [NpO2(OH)2].xH2O was washed twice with H2O, once with acetone and then dried in vacuo. Under an argon atmosphere, the brown residue was suspended in thf (5 mL). A solution of HCl in Et2O (2 mol L−1 < 1 mL) was added until the brown residue dissolved to yield a yellow solution. A few drops of SiMe3Cl were then added to remove any trace of H2O [4c] and the resulting mixture was stirred for 15 minutes to give [NpO2Cl2(thf)]n which was used immediately. A solution of Ph3PNH in THF (29 mg, 0.1 mmol) was added to a solution of [NpO2Cl2] in THF (12.63 mg, 0.053 mmol of 237Np). A yellow precipitate formed upon addition. The solid was left to precipitate and the supernatant was removed. The solid was dried and redissolved in CD2Cl2 and left to stand at room temperature; yellow crystals formed after a few days. 31P{1H} NMR (CD2Cl2): δ 36.1 ppm (s).
Acknowledgments
We acknowledge the Engineering and Physical Sciences Research Council (EPSRC), the School of Chemistry, University of Manchester and ACTINET for funding. In particular, S.M. Cornet was funded from the EPSRC grant no. GR/S95152/01.
Appendix A Supplementary data
CCDC 756667 and 756668 contain the Crystallographic data for compounds 1 and 2 respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via. www.ccdc.ca.ac.uk/data_request/cif or the Cambridge Crystallographic Data Centre. 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +(44) 1223-336-033 or email: deposit@ccdc.cam.ac.uk.