

1 Introduction
La séparation des actinides mineurs et des lanthanides pourrait être une étape importante de la gestion des déchets nucléaires [1]. L’extraction sélective par solvant est le processus le plus souvent envisagé. Son efficacité dépend avant tout de l’aptitude de l’agent complexant à établir des interactions donneur–accepteur avec les ions lanthanides et actinides. Les études menées pendant la dernière décennie ont montré que les ligands N-donneurs aromatiques forment des liaisons chélatantes stables avec, en particulier, les ions lanthanides trivalents [1–4]. L’effet de chélation dépend du nombre d’atomes d’azote capables de former des liaisons avec l’ion métallique à extraire. Ainsi, les ligands tridentate de topographie planaire, tels que 2,2′,6′,2′′-terpyridine (Tpy), 2,4,6-tri-(2-pyridin-2-yl)-1,3,5,-triazine (Tptz), 2,6,-bis([1,2,4,-triazin-3-yl)]pyridine (Btp), etc., sont particulièrement intéressants. Une charge négative importante sur les atomes d’azote dans les cycles aromatiques, pyridine et triazine [5] assure une grande stabilité aux complexes du type [LnL]3+ ou [AnL]3+, qui se forment avec les ions actinides ou lanthanides trivalents dans la phase organique. Cependant des actinides dans leurs états d’oxydation supérieurs (5+) et (6+) peuvent aussi former des complexes stables avec les polypyridines [6–8] comme le montre ce travail, dans lequel l’interaction de Tptz avec Np(V) est abordé via l’étude de composés solides de Np(V).
2 Synthèse et structure cristallographique des composés
Le composé [NpO2Cl(Tptz)H2O]·H2O (I) est obtenu par évaporation lente du mélange contenant les solutions de NpO2Cl dans H2O et Tptz dans CH3OH dans un rapport molaire 1:1. Le composé NpO2Br(Tptz)·n H2O (II) est synthétisé de façon analogue à partir de NpO2Br préparé à partir de NpO2OH et une solution aqueuse de HBr.
Les monocristaux croissent par recristallisation dans le méthanol. Les données structurales ont été obtenues sur un diffractomètre Siemens P3/PC. La structure cristalline a été déterminée par la méthode directe et précisée pour tous les atomes autres que l’hydrogène, en affinant les facteurs d’agitation thermique anisotrope par une procédure des moindres carrés sur le facteur de structure F2. La position de tous les atomes d’hydrogène de la molécule Tptz a été déterminée à partir des cartes de densité électronique.
Dans le composé [NpO2Cl(Tptz)H2O] (I), le polyèdre de coordination (PC) de l’atome de neptunium est bipyramidal, à base pentagonale. Le plan équatorial est formé par les trois atomes d’azote de la molécule Tptz, l’oxygène de la molécule d’eau et l’atome de chlore. Le groupe NpO2 présente une structure pratiquement linéaire et symétrique. Les distances Np–O sont égales à 1,7940(26) Å et 1,8134(26) Å, l’angle O–Np–O est égal à 178,75(12)°. Les distances séparant l’atome de neptunium des atomes équatoriaux ont les valeurs suivantes : Np–N = 2,6045(27)–2,6740(31) Å, Np–O(H2O) = 2,4552(25) Å, Np–Cl = 2,8207(11) Å. Des valeurs analogues de longueur de la liaison Np–N ont été déterminées dans différentes structures : 2,580(5)–2,602(5) Å dans [NpO2NO3(Terpy)H2O] [2] ; 2,617(5), 2,649(5) Å dans [NpO2NCS(Dipy)(H2O)2]·H2O [3] et 2,62(1), 2,66(1) Å dans [NpO2(Dipy)(H2O)3](NO3) [4]. L’écart des atomes au plan moyen équatorial n’excède pas 0,0915 Å (Fig. 1).
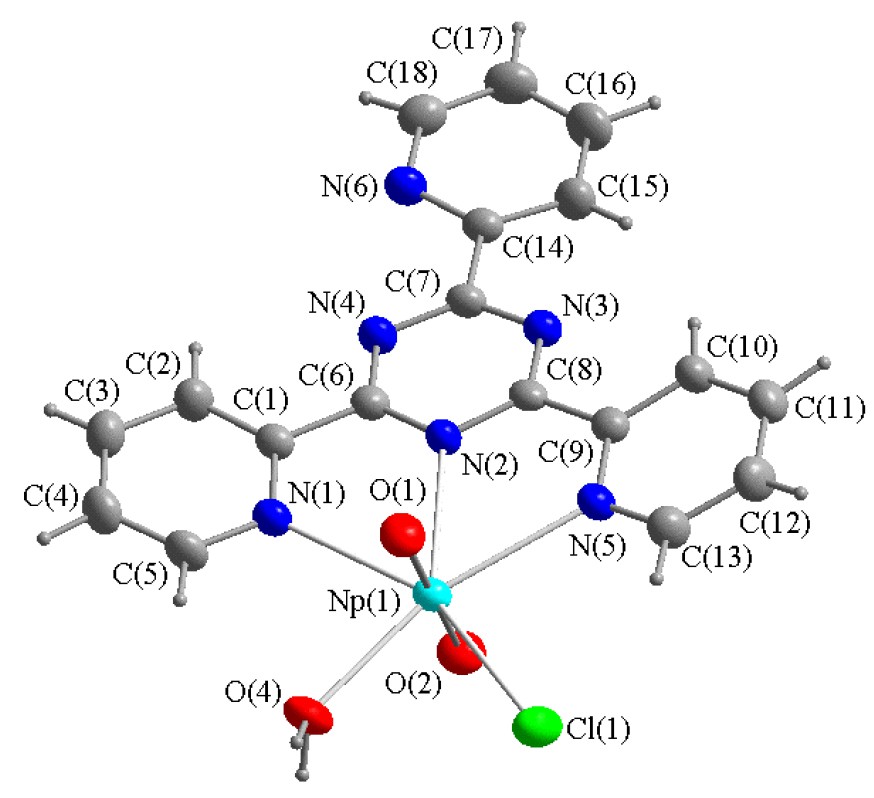
Fragment de la structure de [NpO2Cl(Tptz)H2O]·H2O.
La molécule Tptz, en complexant l’atome de neptunium par sa fonction ligand tridentate, forme dans le plan équatorial du PC de l’atome de neptunium deux metallocycles à cinq atomes. Les cycles aromatiques Py1, Py2, Py3 et triazine ont une structure de type planaire, avec un écart maximum entre atome et plan moyen n’excédant pas 0,0117 ; 0,0103 ; 0,0088 et 0,0120 Å, respectivement. Les angles entre le plan triazine et les plans Py1, Py2, Py3 sont respectivement de 12,4°; 12,0° et 9,7°.
Un système de liaisons hydrogène assure en partie la cohésion de l’assemblage moléculaire. Les paramètres relatifs aux liaisons sont reportés dans le Tableau 1.
Différentes longueurs de liaison (d) et différents angles (ω) dans la structure de [NpO2Cl(Tptz)H2O]·H2O
| Liaison | Longueur, Å | Angle | ω (degré) |
| Np–O(1) | 1,794(3) | O(1)–Np–O(2) | 178,75(12) |
| Np–O(2) | 1,813(3) | O(1)–Np–O(4) | 91,30(11) |
| Np–O(4) | 2,455(3) | O(2)–Np–O(4) | 89,26(11) |
| Np–N(2) | 2,604(3) | O(1)–Np–N(2) | 94,18(11) |
| Np–N(5) | 2,645(3) | O(2)–Np–N(2) | 84,64(11) |
| Np–N(1) | 2,674(3) | O(4)–Np–N(2) | 135,23(9) |
| Np–Cl(1) | 2,8207(11) | O(1)–Np–N(5) | 88,62(11) |
| N(1)–C(5) | 1,337(4) | O(2)–Np–N(5) | 91,18(11) |
| N(1)–C(1) | 1,342(4) | O(4)–Np–N(5) | 162,73(9) |
| N(2)–C(6) | 1,351(4) | N(2)–Np–N(5) | 61,94(9) |
3 Comportement thermique des composés
La décomposition thermique en présence d’air, des composés [NpO2Cl(Tptz)H2O]·H2O (I) et NpO2Br(Tptz)·m H2O (II) s’effectue selon les courbes présentées sur les Figs. 2a,b.
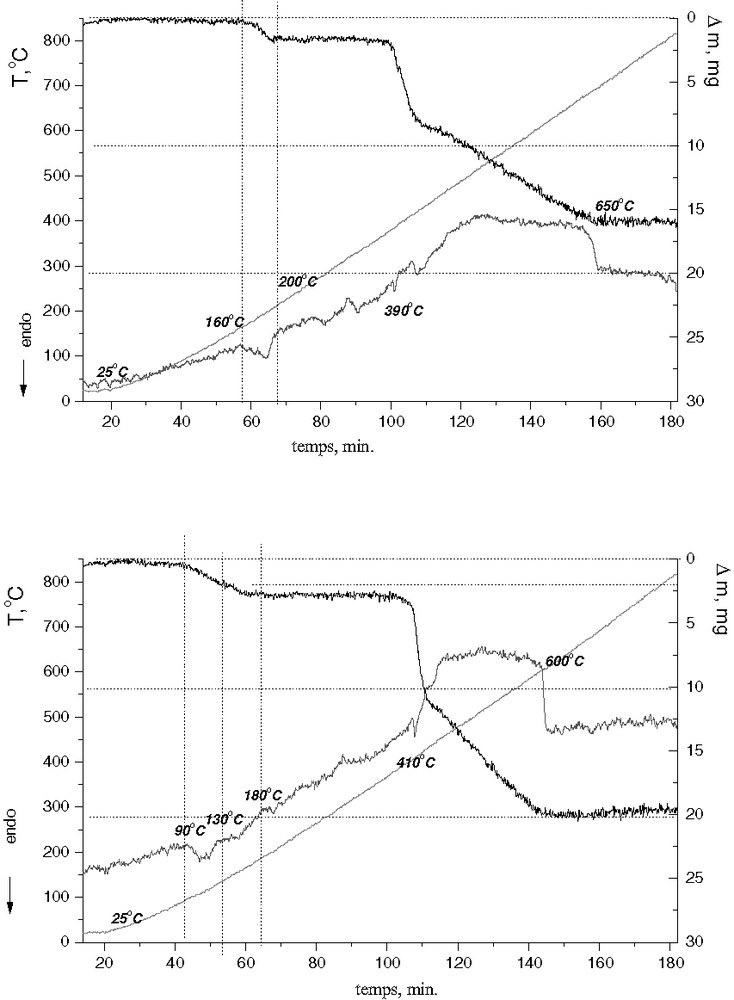
Analyse thermique de [NpO2Cl(Tptz)H2O]·H2O (a) et NpO2Br(Tptz)·m H2O (b). Masse (a) = 26,601 mg ; (b) = 31,511 mg ; vitesse de chauffe : 5 °C min–1.
Pour les composés (I) et (II), la perte totale de masse est de 58,7 et 62,3% respectivement, ce qui correspond à la présence de deux et trois molécules d’eau dans ces composés (m = 3 pour le composé (II)). Les températures du début de déshydratation des composés (I) et (II), sont très différentes. Le composé (I) est stable jusqu’à 160 °C, puis se déshydrate entièrement dans un intervalle de température assez étroit, qui s’achève à 200 °C. Le processus de déshydratation du composé (II) intervient dès 90 °C et s’étend dans un intervalle de température beaucoup plus large, allant jusqu’à 180 °C environ. Cette extension en température est la signature de l’élimination successive des molécules d’eau de cristallisation, qui se traduit par deux effets endothermiques sur la courbe DTA. La température de déshydratation plus élevée du composé chloré (I) correspond à une plus grande stabilité de la liaison avec H2O, comparativement au composé bromé.
Après l’étape de déshydratation, les comportements thermiques de (I) et (II) sont très semblables. La décomposition commence à T = 380 °C (I) et T = 400 °C (II). À 390 °C (I) et 410 °C (II), les effets endothermiques observés correspondent à la réduction de Np(V) en Np(IV), suivie d’une perte rapide de masse de l’échantillon. La décomposition thermique des composés s’achève vers 600 et 650 °C, respectivement.
4 Propriétés spectroscopiques des composés
Les spectres optiques des composés (I) et (II) dans le proche IR, visible et UV ont été relevés sur un mélange comprenant le composé (I) ou (II) et NaCl fondu jouant le rôle de matrice support. Notre pratique de l’étude des spectres d’absorption des composés de Np(V) formés avec des ligands organiques divers a montré que NaCl fondu est à de nombreux égards préférable à KBr : (i) il n’est pas hygroscopique, (ii) il n’entre pas dans des réactions indésirables en phase solide avec la substance étudiée, (iii) il est transparent dans un large domaine spectral. L’utilisation de KBr conduit à la destruction de s-triazine [9], bien que nous ayons pu relever, sous certaines conditions, le spectre IR de s-triazine solide dans KBr et préciser l’attribution de certaines fréquences vibratoires [10].
Les spectres d’absorption dans le domaine spectral UV ont été relevés sur des suspensions huileuses des composés étalées sur des lames de quartz. Il est à noter que les spectres IR du ligand pur, du ligand dans la matrice NaCl, en suspension dans l’huile ou déposé sur un verre KRS-5 sous forme de pellicule par évaporation d’une solution alcoolique sont identiques, montrant que le ligand n’est pas détruit lors de la préparation des échantillons.
Sur la Fig. 3 sont présentés les spectres IR des composés et du ligand pur. Les énergies (cm–1) des principaux modes vibratoires sont les suivantes :
- • Ligand : 3424, 3380, 3262, 3056, 1536, 1468, 1432, 1376, 1260, 1152, 1092, 1048, 1002, 852, 768, 736, 668, 624 ;
- • Composé (I) : 3522, 3432, 3416, 3072, 1626, 1548, 1510, 1468, 1432, 1384, 1372, 1310, 1296, 1260, 1096, 1050, 1031, 1012, 998, 848, 824, 808, 768, 752, 736, 664, 628 ;
- • Composé (II) : 3424, 3360, 3204, 3062, 1636, 1552, 1508, 1432, 1382, 1372, 1258, 1152, 1096, 1052, 1008, 992, 856, 820, 808, 768, 736, 676, 664, 632.
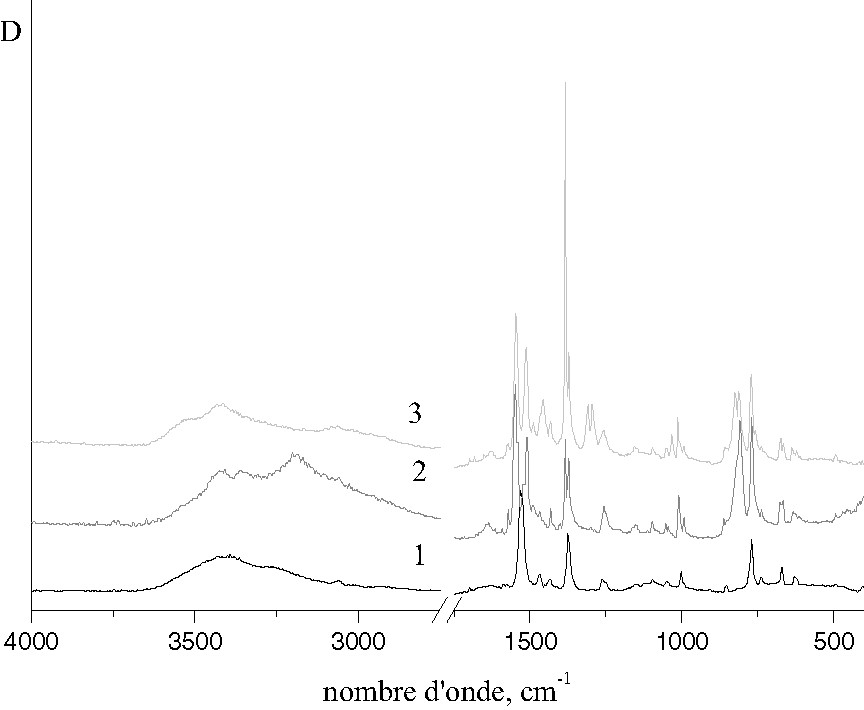
Spectres IR : 1, composé I ; 2, composé II ; 3, ligand pur.
Les spectres de (I) et (II) étant proches dans le domaine d’énergie 500–1600 cm–1, nous pouvons conclure que la coordination de l’ion neptunyl change essentiellement les fréquences de vibration des liaisons C–N, alors que les fréquences correspondant aux vibrations propres de la molécule Tptz restent pratiquement inchangées. Par exemple, la vibration à 768 cm–1 (Ringtors), qui est caractéristique du ligand pur, se retrouve sur les spectres des deux complexes, ce qui indique une faible rigidité de l’assemblage moléculaire.
Les fréquences de vibration de l’ion NpO2+ sont assez proches dans les complexes (I) et (II) : 824,8 et 820,8 cm–1. Elles sont en accord avec un environnement identique dans le plan équatorial de l’ion neptunyl. Cependant, dans le composé (I), on observe un doublet bien résolu, avec une intensité égale des deux composantes, alors que pour le composé (II), la bande d’absorption ν(NpO2+) présente un faible épaulement apparaissant du côté des hautes énergies.
Dans le domaine 4000–3000 cm–1, caractéristique des vibrations ν(OH) et δ(CH), les différences deviennent plus importantes. Elles indiquent la formation de liaisons hydrogène impliquant les molécules d’eau supplémentaires dans la structure du composé (II) et corroborent donc les résultats de l’analyse thermique de ces composés.
La structure identique de la première sphère de coordination de NpO2+ dans les complexes (I) et (II) est confirmée par les spectres d’absorption dans le domaine visible (Fig. 4). Les énergies des transitions électroniques dans la configuration fondamentale 5f2 (λ = 984 nm pour 3H4 → 3F4) ainsi que leur intensité sont très proches (ε = 180 l mol–1 cm–1).
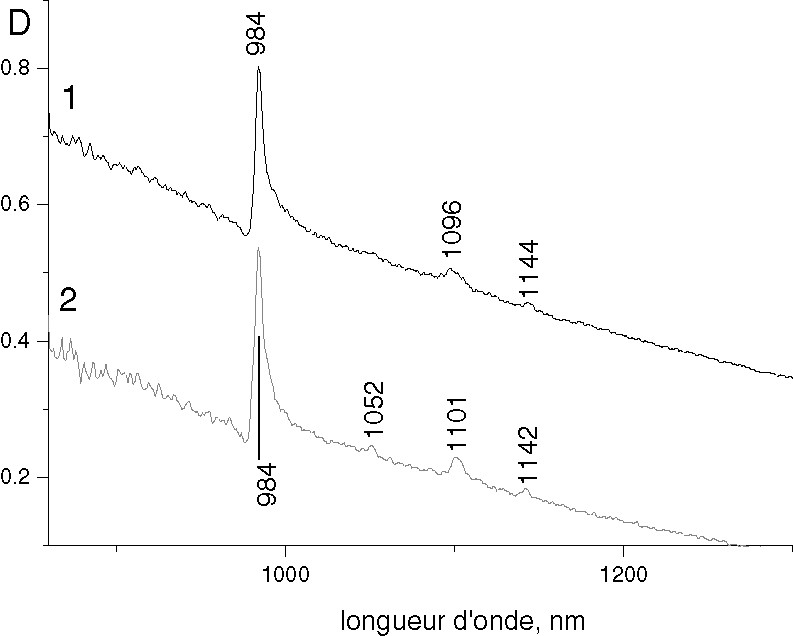
Spectres d’absorption dans le domaine visible : 1, composé I ; 2, composé II.
Dans le domaine UV, on peut observer l’effet de la complexation de l’ion neptunyl par Tptz. Sur la Fig. 5-1, les deux bandes d’absorption à 248 et 292 nm sont caractéristiques du ligand pur [11]. Dans les spectres des complexes (Fig. 5–2,3), ces bandes se déplacent vers les faibles énergies en s’élargissant, alors que leur intensité diminue. Ce comportement est caractéristique de la formation d’un complexe.
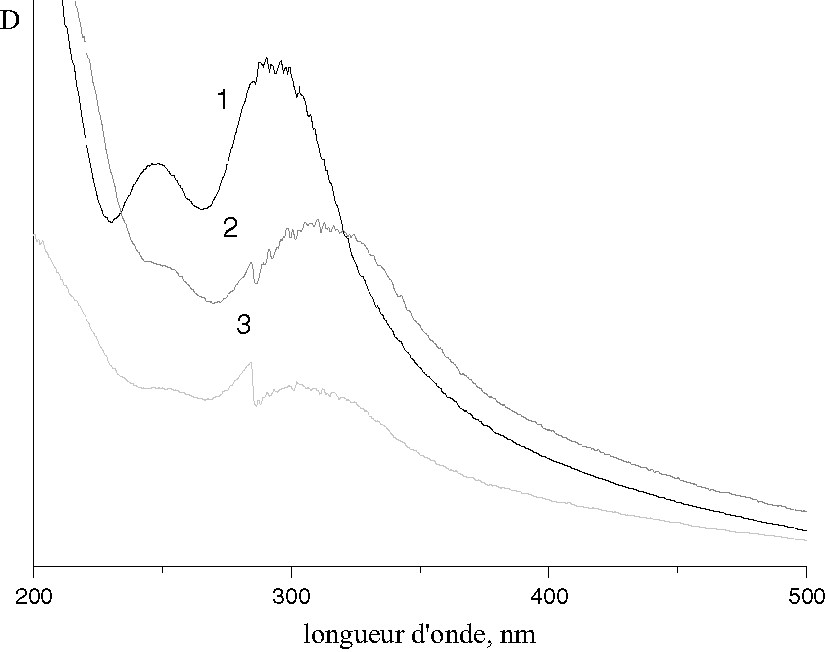
Spectres d’absorption dans le domaine UV : 1, ligand pur ; 2, composé I ; 3, composé II.
L’influence de la nature de l’anion (Cl– ou Br–) sur leur position énergétique est très faible et difficile à apprécier.
5 Conclusion
Ce travail entre dans le cadre d’une étude systématique des complexes d’ions actinide formés avec des ligands azotés via la synthèse de composés solides. Les distances inter-atomiques ion actinide–atome d’azote, accepteurs ou donneurs d’électrons, sont des paramètres critiques qui déterminent le caractère covalent des liaisons assurant la stabilité des complexes. Ces données cristallographiques sont donc essentielles. Elles permettent aussi de tester les simulations ab initio qui tentent de prévoir les molécules complexantes les plus performantes pour séparer efficacement les ions actinide/lanthanide dans un même état d’oxydation.
Cette étude met en évidence le rôle différencié, clairement démontré dans [5], que jouent les ions azote latéraux N(1) et N(5) et central N(2) lors de la formation du complexe de Np(V) avec le ligand polyazoté Tptz.
Remerciements
Ce travail a été réalisé grâce au soutien financier du Fonds russe de recherches fondamentales (bourse n° 03-03-32649).