

1 Introduction
The chemistry of plutonium (Pu) has important applications to civilian nuclear fuel production, spent nuclear fuel processing, nuclear weapons production/stewardship, nuclear waste disposal, and environmental monitoring [1(and references therein),2]. Of particular interest is electricity production from nuclear fuels, which is prompting new reactor designs, as well as research on new fuel and waste forms. Application areas emphasize the need for flexible and efficient separations within processes that recycle used nuclear fuels to minimize the actinide content, volume, and radiotoxicity of waste that will require long-term monitoring or disposal, and to encapsulate the radioisotopes within extremely robust forms [2,3]. An understanding of actinide coordination chemistry, and a broad range of complexes that can underpin advanced and closed nuclear fuel cycles. The opportunity is great, particularly for the chemistry of the lighter transuranic elements (Np-Cm), to move relatively quickly from research to scale up and testing for application.
The general approach to designing complexes that may be used to model separations agents is to first understand the fundamental reasons (detailed bonding, electronic structure, solvation, etc.) that known extractants exhibit the selectivity that they do, and then to extend studies to a wider range of ligands that can be used in similar or new processes. For example, dithiophosphinic acids display very high separation factors for An(III) vs. Ln(III) ions [4–6]. Lanthanide and actinide complexes with other underexplored “soft” donor atom ligands can test hypotheses for the observed selectivity and create a reservoir of basic electronic and bonding knowledge for molecular chemistry. Advances in the chemistry of Np, Am, and Cm carry similar importance, but are outside the focus of this plutonium-oriented paper.
While the research needs are significant, progress in plutonium studies trails far behind contemporary comprehension of transition metal, lanthanide, and early actinide (thorium (Th), uranium (U)) chemistry. The paucity of fundamental science advances, as well as applications, is partly due to the special radiological facilities and monitoring programs necessary to acceptably work with α-emitting transuranic radionuclides (that have higher specific activity than the naturally available isotopes of Th and U). Specifically, 239Pu is the most abundant Pu isotope with a half-life of 24,110 years, and it is fissile. It is therefore difficult to acquire sufficient quantities of Pu isotopes for laboratory-scale studies and few forms are available, even to researchers with access to secure, appropriately equipped facilities. Academic programs that research transuranic chemistry are now generally using trace quantities of plutonium and focusing more studies on surrogates, which are inherently limited. (Importantly, a few universities do still have, or have recently developed, the radiological facilities to perform small-scale transuranic synthetic work, primarily with 237Np and 242Pu. Their research is essential to advance the field.) A brief description of the complexities of plutonium research is included in our overview, because research in transuranic chemistry is limited and it is valuable to disseminate information that is common knowledge for the chemistry of most other elements.
What can nonaqueous chemistry teach us about Pu chemistry? Most aqueous-stable ligands tend to yield complexes where actinide–ligand interactions are predominately ionic in nature. (The obvious exception is the AnO2n+, n = 1 or 2, An = U, Np, Pu, Am, actinyl moeity, ions that contain inherent metal-oxygen multiple bond character). However, the selectivity of certain soft donor ligands (e.g., N and S) for An(III) vs. Ln(III) ions is hypothesized to stem from a combination of modest enhancement of covalency in actinide-soft donor bonding vs. lanthanide-soft donor bonding [4–7] and differences in solvation [5]. The observed separations, and causal effect of covalency, have been reported since the discovery of many of the actinide isotopes in the 1950s [8]. The extent and potential amplification of the effects for applications, as well as the nature of the metal-based orbitals that interact with ligand-based orbitals, are active areas of research. Moving to nonaqueous solvent regimes, in the absence of moisture and oxygen, opens up access to a broad range of ligands that can be utilized to understand and quantify detailed bonding information, including the relative participation of actinide valence 6d and 5f orbitals. Nonaqueous experiments also offer conditions under which ligand composition and solvent environment can be tailored and systematically modified, much more readily and over a broader range compared with aqueous systems. Relevant to our specific research, homoleptic complexes allow for focus on a single, or a small number of bond types in a large coordination sphere. This type of plutonium complex has several of the same type of bond, and the covalency is distributed throughout the coordination sphere. Conversely, stabilizing scaffolds (e.g., the cyclopentadienyl anion and its derivatives) are employed to isolate complexes/bond types that localize covalency, and potentially extreme reactivity, in one or two sites. Far more research has been done using the latter approach. We therefore have focused on expanding the former set and expanding the range of characterized compounds and types of chemical bonds.
The impressive range and unexpected reactivity discovered in nonaqueous and organometallic uranium chemistry offers a glimpse of how transuranic research in the nonaqueous realm has the potential for significant impact [1,9]. One indicator of the current knowledge gap between uranium and the transuranic ions is exemplified by entries in Cambridge Crystallographic Database (CSD) [10]. Currently, there are 2578 entries that contain U, 117 that contain Np, 53 that contain Pu (Fig. 1), and only 12 molecules containing transplutonium metal centers. Clearly, there is great opportunity to increase our knowledge of the structural preferences, reactivity signatures, electronic structure, and bonding interactions in nonaqueous Pu complexes and to close the knowledge gap with U chemistry.
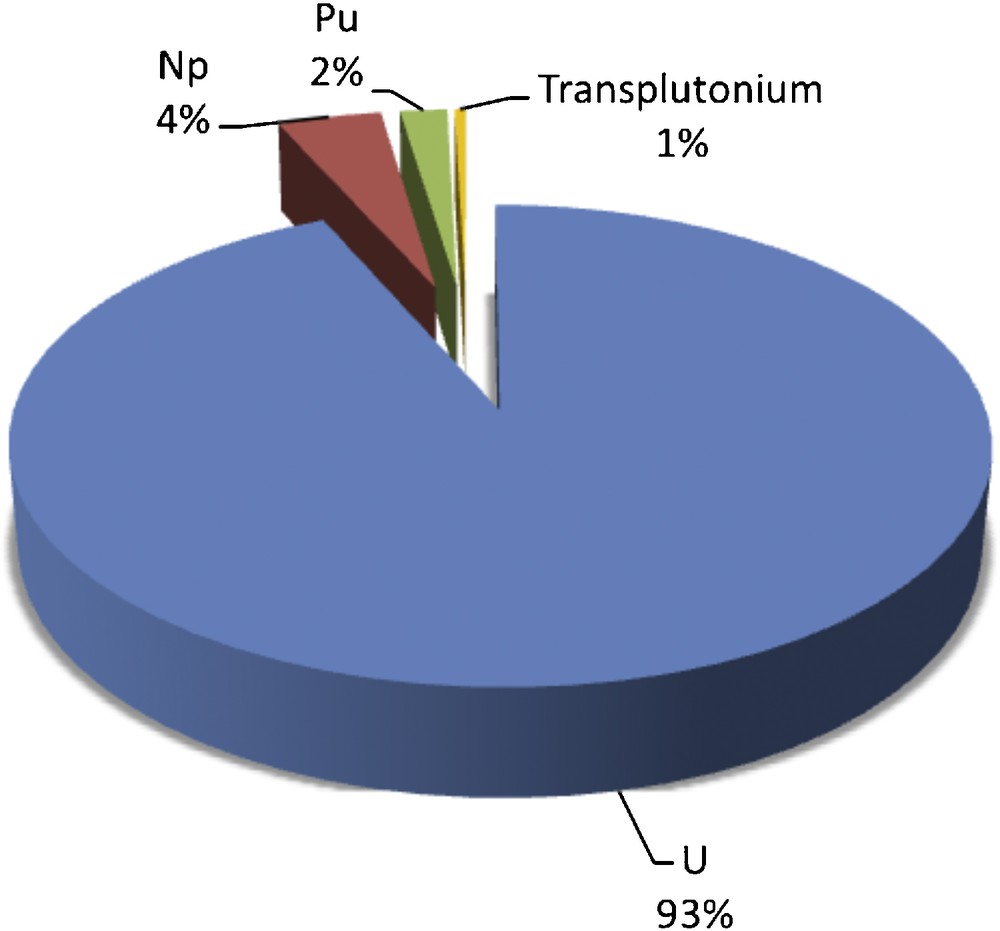
The number of entries in the Cambridge Structural Database containing different actinide elements as % of the total actinide entries.
In the interest of brevity, we focus this account to research efforts in nonaqueous Pu chemistry, performed in the absence of air and moisture, in our laboratory. Several international researchers are currently engaged in elegant and insightful related or comparable studies, many of which are contributing to this same journal issue.
2 Literature precedent in nonaqueous Pu chemistry
We begin by very briefly summarizing the excellent prior work, mostly conducted between 1952 and 1985, which paved the way for our nonaqueous plutonium chemistry research. More detailed reviews can be found in the recent volumes of ‘The Chemistry of the Actinide and Transactinide Elements’ [1].
The [PuIVCl6]2− anion, as various salts, including the organic soluble [Et4N]+ and [Me4N]+, has provided a useful entry to a number of early Pu(IV) complexes [11]. In 1957, Bradley et al. prepared Pu(IV) alkoxide complexes of general formula Pu(OR)4 [12]. Amide and phosphine oxide adducts of general formula PuCl4L2 and PuCl4L3 (L = amide or phosphine oxide) were reported by Bagnall and coworkers by employing Cs2[PuCl6] as the precursor [13]. The earliest plutonyl nonaqueous complexes came from Staritzky and Singer in the form of [Et4N]2[PuO2Cl4] and [Me4N]2[PuO2Cl4] [11]. The first organometallic Pu molecule appeared in 1965 when Baumgärtner detailed the synthesis of Pu(C5H5)3 through transmetallation of molten (C5H5)2Be at 70 °C with PuCl3 [14]. An alternative larger scale route to Pu(C5H5)3 using (C5H5)2Mg was developed in 1974 [15]. Syntheses of other Pu molecules containing cyclopentadienyl (Cp) ligands have since been achieved with (C5H5)3Pu(thf), (C5H5)3PuCl, (C5H5)3Pu(NCS), [AsPh4][(C5H5)3Pu(NCS)2], (C5H5)PuCl3L2 and (C5H5)Pu(NCS)3L2 (L = amide or phosphine oxide) all reported [16]. Following the seminal discovery of uranocene and then Pu(C8H8)2 in 1970 [17], a number of cyclooctatetraene (COT) and derivative complexes were published with Pu(EtC8H7)2, Pu(nBuC8H7)2, Pu(tBuC8H7)2, [K(thf)2][Pu(RC8H7)2] (R = alkyl or H), [K(diglyme)][Pu(C8H8)2], and Pu(1,3,5,7-Me4C8H4)2 (which was actually shown to be [K(thf)2][Pu(1,3,5,7-Me4C8H4)2]) all synthesized [18]. Other nonaqueous Pu complexes that do not incorporate Cp or COT type ligands are the borohydride Pu(BH4)4 [19], the dithiocarbamate Pu(S2CNEt2)4 [20], and the organic solvates of Pu halides PuI3(thf)x and PuI3L4 (L = thf, pyridine, dmso). The synthetic utility of those organic soluble halides was demonstrated through the preparation of Pu[N(SiMe3)2]3, Pu(O-2,6-tBu2C6H3)3, Pu[CH(SiMe3)2]3, Pu(O-2,6-tBu2C6H3)3L (L = triphenylphosphine oxide, 4,4′-dimethoxybenzophenone, N,N-diisopropylbenzamide), and Pu(OiPr)3 [16e,21–24].
Of note is that entries in the CSD corresponding to the above nonaqueous compounds are restricted to just six compounds: [Et4N]2[PuCl6], [Me4N]2[PuCl6], [Et4N]2[PuO2Cl4], [Me4N]2[PuO2Cl4], Pu(Et2NCS2)4, [K(solvent)][Pu(C8H8)2] (solvent = diglyme or 2 thf molecules). However, these data were generated before the advent of ‘routine’ single-crystal X-ray structure determinations and are confined to lattice constants, and 3-dimensional coordinates for the atoms in the structures are not available. Jensen and Almond recently reported the full single crystal structure of Pu(Et2NCS2)4 [25], providing more detailed characterization of the dithiocarbamate plutonium complexes.
3 Recent nonaqueous plutonium coordination complexes prepared at Los Alamos National Laboratory
Our approach over the last several years has been to study in detail nonaqueous Pu complexes to address specific questions regarding the nature of actinide bonding to soft donor atom ligands. Our aims are to examine bonding trends across the actinide series, compare them to analogous lanthanide complexes, and understand how oxidation state impacts the reactivity and bonding properties. Within this strategy, we focus on syntheses of homoleptic complexes allowing ‘isolated’ analyses of the soft donor bond of interest across a large coordination sphere (compared to many other studies in which there are different metal-ligand bond types with the soft-donor bond of interest occupying a fraction of the coordination sphere). These types of compounds are also more directly useful to designing extractants for separations applications. Another aim is to probe the limits of hard 5f metal–soft ligand interactions, by targeting complexes with ‘ultra-soft’ ligands.
3.1 Practicalities of nonaqueous plutonium chemistry
It is useful to briefly describe general experimental environments and methods, so that the reader can gain insight as to how transuranic research differs from uranium, thorium, and non-radioactive research. Our air-sensitive transuranic chemistry is conducted inside a helium atmosphere glove-box operated at a slight negative pressure, with inlets and outlets equipped with High Efficiency Particulate in Air (HEPA) filters to contain radioactive particulates. The exhaust gasses are vented into a HEPA-filtered fumehood, intended to further prevent release of transuranic isotopes. Laboratory surfaces, equipment, air, and personnel, are frequently monitored for contamination. Samples are carefully measured, packaged, and tracked when they are released from specific areas. Most practices common in synthetic laboratories are done quite differently within radiologically-controlled areas. For example, researchers wear respirators and multiple layers of gloves and lab clothing when performing maintenance tasks that open the dry box (e.g., glove changes, vacuum pump changes). Reactions are typically performed on a scale of 10 mg of 239Pu, and a researcher will typically use less than 500 mg of 239Pu in dry-box experiments over a year. Due to restrictions, experiments are planned in detail well in advance (i.e., if a synthetic idea has not already been shown to be successful and optimized for other f elements then one risks a careless waste of a precious resource). Starting materials are limited to plutonium solutions in multimolar HCl or HNO3, α or δ phase metal (Fig. 2), or oxide, PuO2. Although performed on a small scale, complexes are comparable to those of other metal ions in terms of their range of morphology, structures, and bulk appearance (Fig. 2).

Plutonium starting material, α phase metal (left) and molecular complexes, [PuO2Cl2(thf)2]2 (center) and Pu[N(SePPh2)2]3 (right, structure is shown in Fig. 9).
Chemists working with transuranic compounds are challenged to develop multiple-containment strategies to collect spectroscopic and analytical data on samples while preventing personnel and equipment exposure to α radioactivity. For example, single crystals containing Pu are loaded into a capillary using a microscope connected to an external video monitor and sealed with wax inside the dry-box, then further sealed with an acrylic coating to provide structural integrity to the capillary. The sample is then monitored and packaged for transport to the diffractometer. The whole process will typically take a day. Similarly, transuranic samples for NMR experiments are placed inside PTFE liners that are then placed inside glass NMR tubes. Very detailed procedures, processes, and policies have been developed for X-ray absorption, ESR, electrochemical, and other analyses. In our experience, the multiple containment and methods used to prevent contamination during sample characterization result in roughly 10 times the experimental time compared to that needed for a non-transuranic metal compound.
3.2 Organic soluble precursors
The first challenge to a wide-ranging study is the lack of well-defined organic soluble nonaqueous Pu molecules that can be readily prepared under relatively non-hazardous conditions (avoiding the use of high temperatures, high pressures, toxic gases, or highly volatile liquid reagents) and employed as synthetic precursors.
In 1994, Avens et al., elegantly demonstrated that a simple route to organic soluble U(III) halide adducts from uranium metal proceeds even more cleanly to prepare Pu(III) halide adducts from Pu metal with iodine in coordinating solvents (thf or pyridine) [22]. PuI3(thf)4 was treated with Na[N(SiMe3)2] to yield the silyl amide complex Pu[N(SiMe3)2]3, itself a useful synthon. No molecular structures were obtained on these compounds. We repeated the syntheses and isolated crystals from a thf solution of PuI3(thf)4 at −35 °C. The single crystal X-ray diffraction data confirm the compound is isostructural with the uranium analog, UI3(thf)4, with the seven coordinate Pu center displaying a distorted pentagonal bipyramidal geometry (Fig. 3) [26]. We also determined that careful control of iodine stoichiometry was important to avoid formation of [PuI2(thf)5][I3] as an unintended product. Oxidation of Pu metal with bromine in thf led to PuBr3(thf)4 (Fig. 4), which was structurally identical to the PuI3(thf)4 molecule [26]. Through a modified synthetic procedure, we isolated and structurally characterized Pu[N(SiMe3)2]3, which as expected is isostructural to the uranium analog with the metal atom occupying pyramidal geometry (Fig. 5) [27]. Comparison to the Ce(III) analogue showed an interesting difference between the agostic interaction distances, with the Ce–C distance being 0.138 Å longer than the corresponding Pu–C distance. Seeking alternatives to the Pu(III) halide precursors, Pu metal was oxidized with either AgPF6 or TlPF6 in acetonitrile to yield the homoleptic complex [Pu(MeCN)9][PF6]3·MeCN [28]. Turning to Pu(IV), apart from salts of [PuCl6]2−, no other organic soluble precursors were available to begin to explore nonaqueous tetravalent plutonium complexes. Indeed, our attempts to prepare Pu(IV) precursors led to other products as we discuss later (section 3.4). [Et4N]2[PuO2Cl4] and [Me4N]2[PuO2Cl4] provide the only literature precedent of organic soluble plutonyl molecules with potential suitability for synthetic exploration [11]. To open up new synthetic routes, we began to take advantage of plutonium materials that can be isolated from acidic aqueous stock solutions. The inorganic carbonates of actinide ions are readily prepared by precipitation via the addition of CO2(g) or common carbonate salts. For example, precipitation of PuO2CO3 from aqueous Pu(VI) solution followed by dissolution with HCl/Et2O in thf led to the isolation of [PuO2Cl2(thf)2]2 as the first structurally and spectroscopically characterized anhydrous plutonyl(VI) molecule (Fig. 6) [29]. This complex should readily undergo metathesis and Lewis base displacement reactions. Preliminary reactivity studies with triphenylphosphine oxide (TPPO) exhibited the expected Lewis base displacement. We are not aware of any plutonyl(V), PuO2+, nonaqueous precursors, although careful one-electron reduction of a suitable PuO2Cl2 adduct may be a promising avenue to explore.
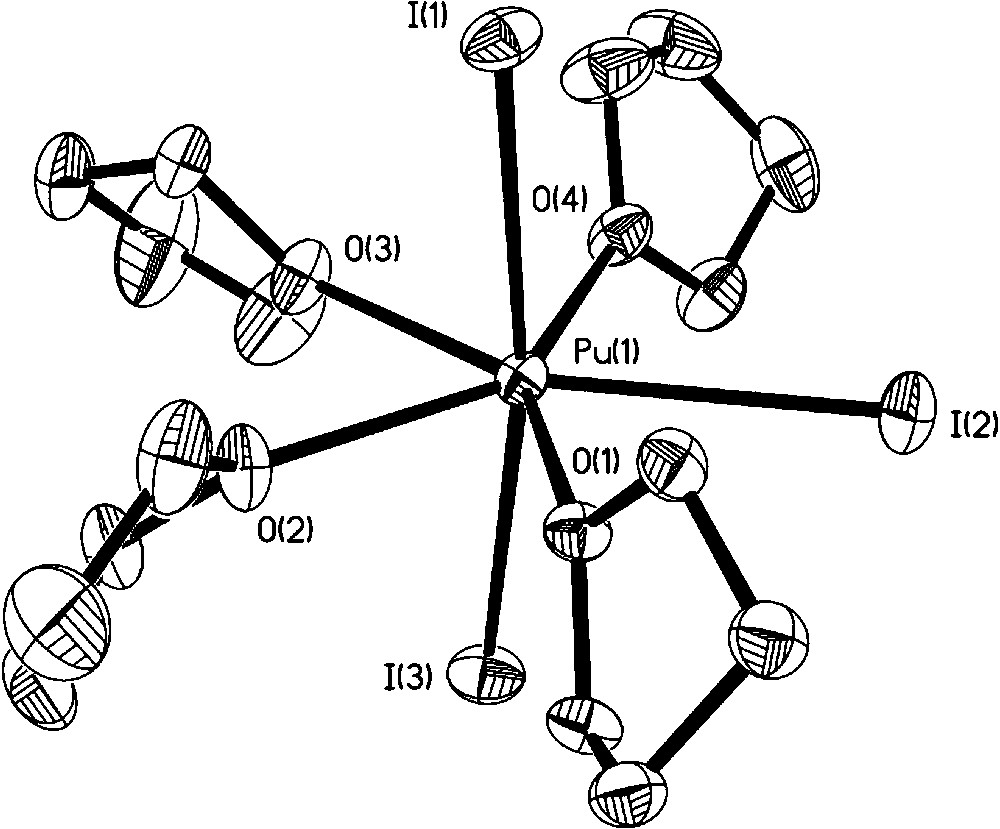
Molecular structure of PuI3(thf)4.
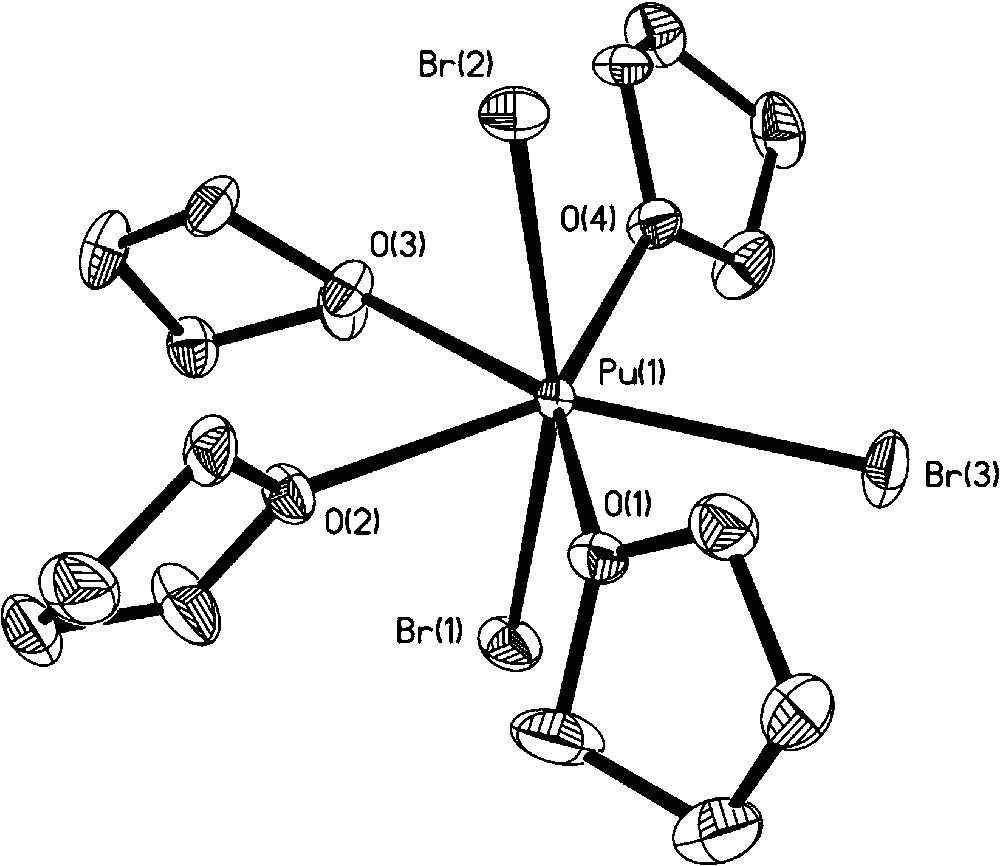
Molecular structure of PuBr3(thf)4.
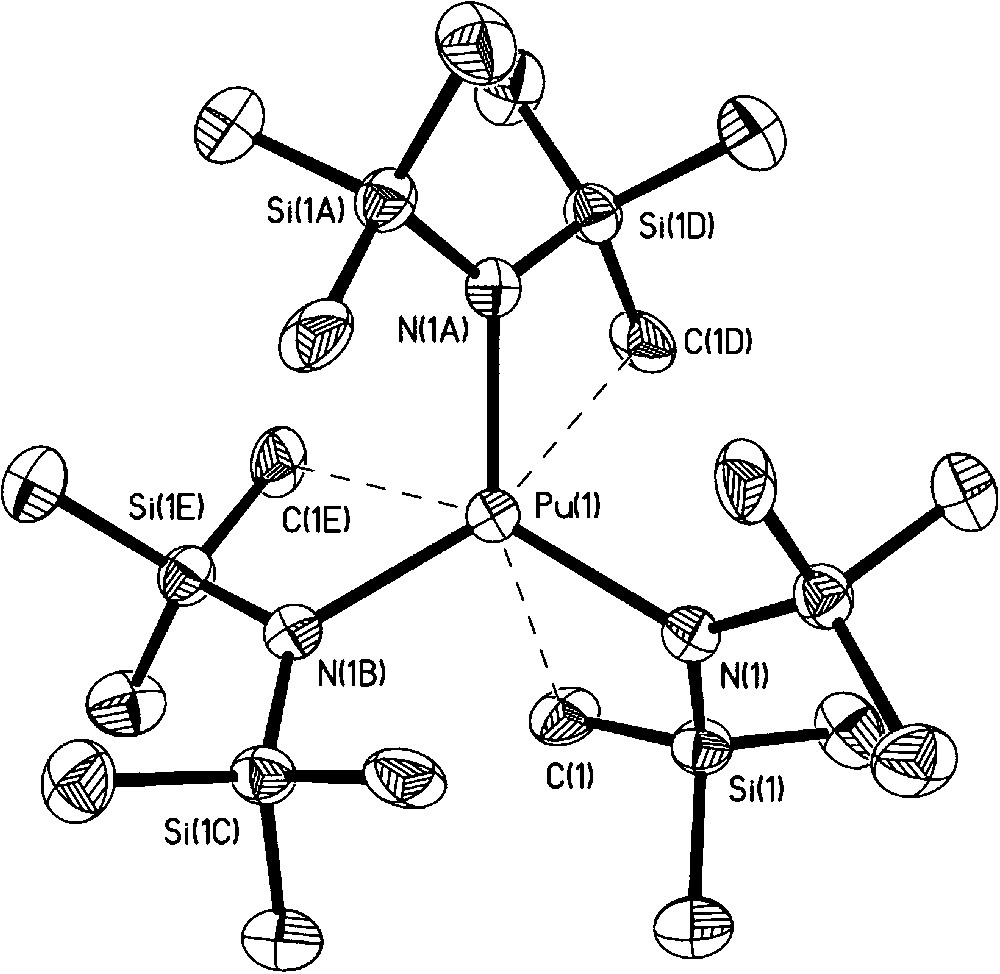
Molecular structure of Pu[N(SiMe3)2]3.
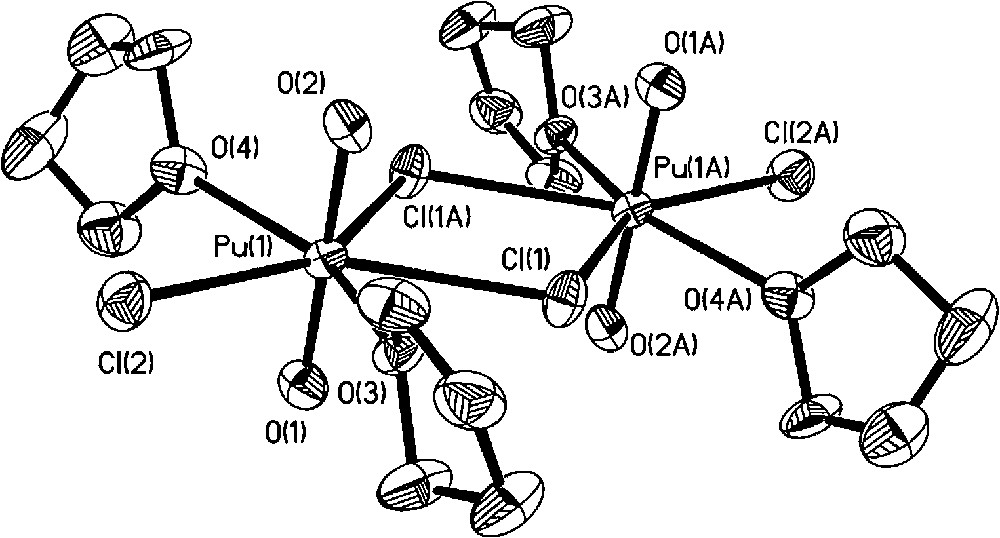
Molecular structure of [PuO2(thf)2Cl2]2.
3.3 Trivalent plutonium complexes with soft donor ligands
With relevance to understanding separation behavior, specifically the difference in actinide and lanthanide distribution coefficients from aqueous solution into organic solvents that contain soft-donor chelates, several groups are researching covalency differences between the 4f and 5f series compounds. Our approach has initially focused upon analyzing differences between An(III)-L and Ln(III)-L bond lengths within single-crystal X-ray diffraction studies of isostructural soft donor complexes as an indicator of covalency differences. Experiments and analysis are being expanded to include modeling, electronic structure theory, and the set of modern spectroscopic techniques (magnetic, optical, vibrational, X-ray absorbance, etc.) to understand the electronic structure and bonding that comprise “covalency”.
The premise is that if 4f and 5f metal ions of near identical ionic radii are compared, then significant discrepancies in bond lengths are unlikely to be attributable to ionic factors. For example, upon comparing a U(III) and Ce(III) phosphite complex, the U–P distance was found to be 0.098 Å shorter than the corresponding Ce–P distance [30]. There are also various examples of comparisons of U–L to La–L distances with N and S donors that have revealed a shorter U–L length up to the order of approximately 0.05 Å (See, for example [31,32], and references therein). It is important to pursue such comparisons for Pu–L vs Ln–L bond lengths to be able to understand the effect of the actinide contraction upon bonding differences, compared to other actinides and also lanthanides of similar ionic radii. Developing bonding trends across the 5f series also supports the link to separations application because it is the transuranic elements (Np, Pu, Am, Cm) that pose the most difficult separation challenges in the nuclear industry.
In 2005, we synthesized two soft donor Pu(III) complexes [33]. Firstly, oxidation of Pu metal with I2 in acetonitrile with subsequent addition of tpza (tpza = tris([2-pyrazinyl)methyl]amine) afforded crystals of Pu(tpza)I3(MeCN) that were structurally characterized (Fig. 7). The Pu metal is eight coordinate and occupies a distorted square anti-prismatic geometry. There is no isostructural lanthanide complex for comparison but in the related Nd(III) complex, [Nd(tpza)(CH3CN)3(H2O)3][ClO4]3, the Nd–N(amine) distance is longer than the Pu–N(amine) distance by 0.116 Å. Secondly, rather than adding tpza, if the cyclic thioether 9-ane-S3 is added to the solution of Pu(III) in acetonitrile then single crystals of Pu(9-ane-S3)I3(MeCN)2 can be grown (Fig. 8). This is the only example of a neutral S donor ligand coordination to a Pu metal center. The average Pu–S distance is 0.028 Å shorter than the average U–S distance in the isostructural U(III) compound, consistent with the actinide contraction and higher positive charge density of Pu(III) compared to U(III). No analogous Ln(III) compound of comparable ionic radii has been reported.

Molecular structure of Pu(tpza)I3(MeCN).
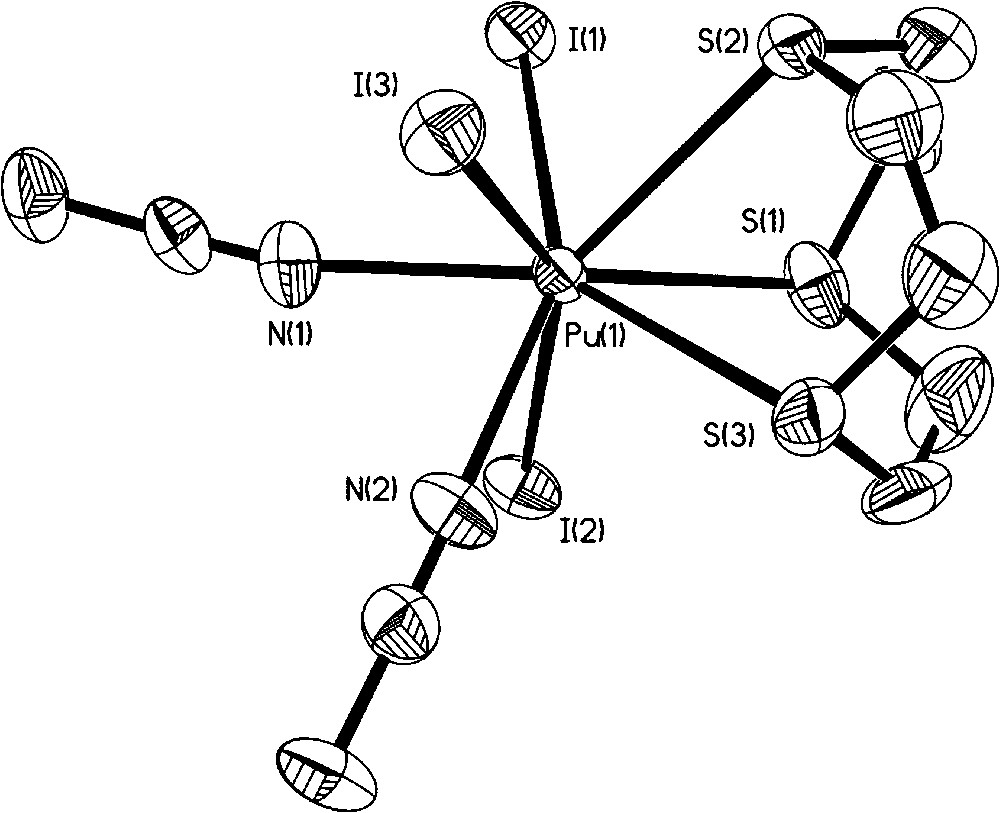
Molecular structure of Pu(9-ane-S3)I3(MeCN)2.
Having established the viability of preparing soft donor Pu complexes under inert atmospheric conditions, a set of molecules were synthesized to probe soft donor bonding in a systematic and detailed manner. Previous uranium based experiments tended to be restricted to heteroleptic examples compared to either La(III) or Ce(III) complexes and with no systematic variables that could be tuned. In our experiments, we decided to compare U(III) vs La(III), and Pu(III) vs Ce(III) as the closest matched “pairs” based upon ionic radii published by Shannon [34]. Choosing two different actinide ions and lanthanide ions and attempting to isolate a series of isostructural complexes would allow the effect of the 5f and 4f contractions upon bond length comparisons to be assessed. For a suitable ligand class, we identified imidodiphosphinochalcogenides of general formula [N(EPR2)2]− as having the potential to fulfill our intended criteria. The identity of the E atom can be changed from S to Se for R = Ph, and from S to Se to Te for R = iPr, providing a mechanism to assess the effect of introducing progressively softer donors into the same ligand architecture. The ability to incorporate different R groups into the same ligand allows investigation of steric factors upon the bonding. We also anticipated that this ligand class would be amenable to the isolation of homoleptic metal complexes lending a focus on only the soft donor bonds of interest. Treatment of Pu[N(SiMe3)2]3 with NH(EPPh2)2 (E = S, Se) afforded the homoleptic complex Pu[N(EPPh2)2]3 in which both the E atoms and N atom of all three ligands make up a nine coordinate Pu atom in distorted tricapped trigonal prismatic geometry (Scheme 1 and Fig. 9 for example structure) [35]. A similar reaction with NH(EPiPr2)2 also yields a homoleptic complex, Pu[N(EPiPr2)2]3, but the increased steric requirements of iPr compared to Ph forces the Pu center to adopt a six coordinate distorted trigonal prismatic geometry in which only Pu–E bonds are present (the N atom does not coordinate). In the case of the Te donor ligand, PuI3(py)4 was treated with [Na(tmeda)][N(TePiPr2)2] to afford Pu[N(TePiPr2)2]3 after workup (Scheme 2 and Fig. 10) [35]. Comparison of the Pu complexes to analogous Ce molecules, and U comparisons to La, unveiled a consistent trend of shorter An–E bonds compared to Ln–E bonds in all cases (Tables 1 and 2). The largest difference was observed between U[N(TePiPr2)2]3 vs La[N(TePiPr2)2]3 (0.060 Å), and Pu[N(TePiPr2)2]3 vs Ce[N(TePiPr2)2]3 (0.059 Å), as might be predicted since Te is a softer donor than S or Se. Perhaps somewhat surprisingly, the magnitude of the bond length differences between U–E vs La–E and Pu–E vs Ce–E are essentially identical instead of decreasing across the f-element series. This finding poses intriguing questions about what might be discovered experimentally for these systems as far across the 5f series as Am(III) and Cm(III).
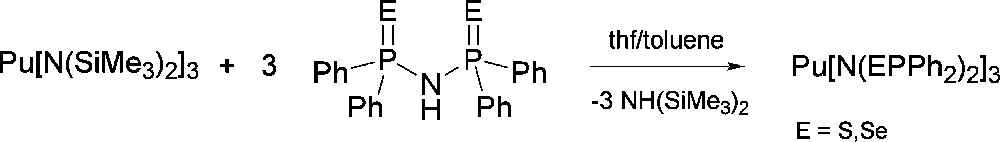
Synthetic route to Pu[N(EPPh2)2]3 complexes.
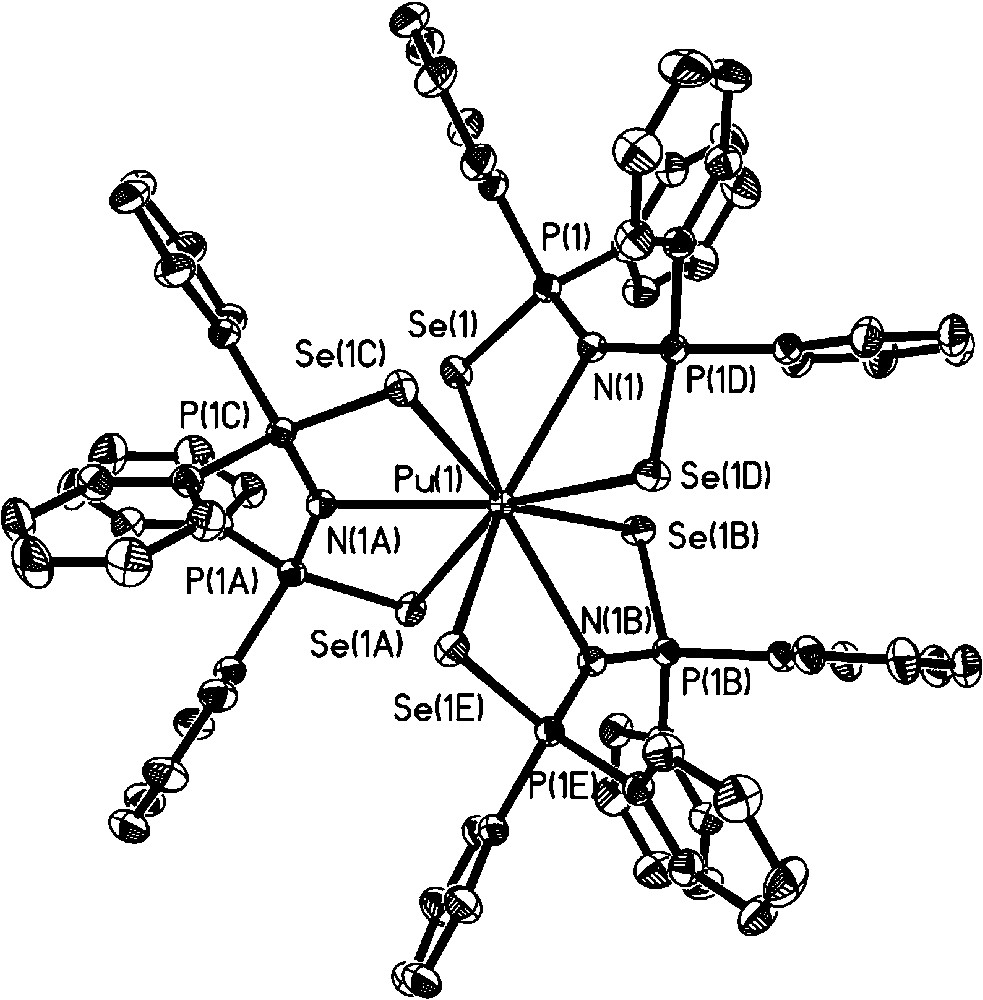
Molecular structure of Pu[N(SePPh2)2]3.
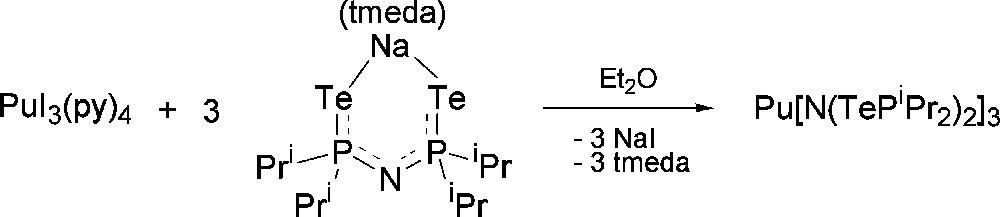
Synthetic route to the Pu[N(TePiPr2)2]3 complex.
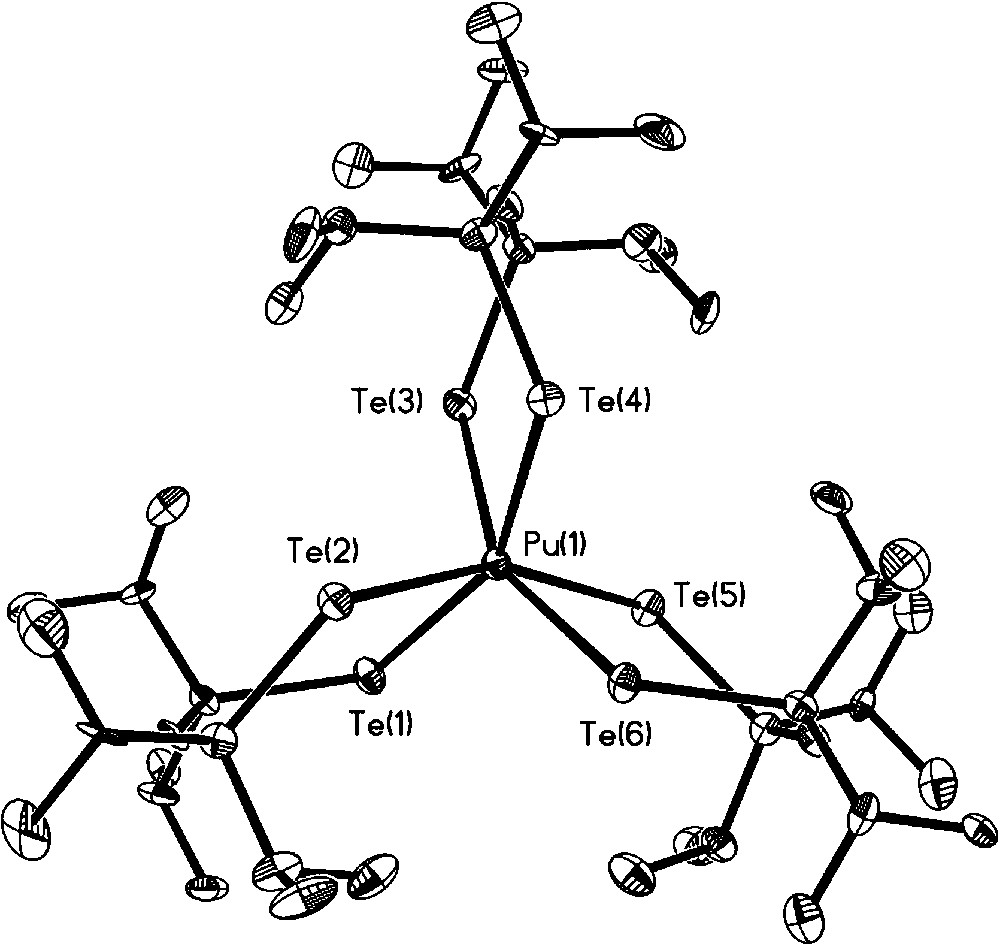
Molecular structure of Pu[N(TePiPr2)2]3.
Comparison of M–E bond distances in M[N(EPPh2)2]3 complexes.
| Compound | Bond | Distance | Difference |
| U[N(SPPh2)2]3 | U–S | 2.9956(5) | – |
| La[N(SPPh2)2]3 | La–S | 3.0214(11) | 0.026(1) |
| Pu[N(SPPh2)2]3 | Pu–S | 2.9782(6) | – |
| Ce[N(SPPh2)2]3 | Ce–S | 3.0052(6) | 0.0270(8) |
| U[N(SePPh2)2]3 | U–Se | 3.0869(4) | – |
| La[N(SePPh2)2]3 | La–Se | 3.1229(3) | 0.0360(5) |
| Pu[N(SePPh2)2]3 | Pu–Se | 3.0710(2) | – |
| Ce[N(SePPh2)2]3 | Ce–Se | 3.1013(3) | 0.0303(4) |
Comparison of M–E bond distances in M[N(EPiPr2)2]3 complexes.
| Compound | Bond | Average distance | Difference |
| U[N(SPiPr2)2]3 | U–S | 2.854(7) | – |
| La[N(SPiPr2)2]3 | La–S | 2.892(1) | 0.038(7) |
| Pu[N(SPiPr2)2]3 | Pu–S | 2.819(3) | – |
| Ce[N(SPiPr2)2]3 | Ce–S | 2.864(2) | 0.045(4) |
| U[N(SePiPr2)2]3 | U–Se | 2.964(7) | – |
| La[N(SePiPr2)2]3 | La–Se | 3.019(3) | 0.055(8) |
| Pu[N(SePiPr2)2]3 | Pu–Se | 2.917(4) | – |
| U[N(TePiPr2)2]3 | U–Te | 3.164(2) | – |
| La[N(TePiPr2)2]3 | La–Te | 3.224(3) | 0.060(4) |
| Pu[N(TePiPr2)2]3 | Pu–Te | 3.123(3) | – |
| Ce[N(TePiPr2)2]3 | Ce–Te | 3.182(1) | 0.059(3) |
The series of structurally characterized lanthanide and actinide(III) imidodiphosphinochalcogenides are now being spectroscopically analyzed to provide detailed information on the molecular bonding, thermodynamic stability and physicochemical properties. For example, multinuclear NMR measurements of paramagnetic U(III) and Pu(III) molecules reveal the relative influence of the actinides with 5f3 and 5f5 electronic configurations on through bond and through space coupling with ligand-based spins. The 31P NMR shifts in Pu[N(EPR2)2]3 (R = Ph or iPr) complexes in deuterated organic solvents range from −1.4 ppm for Pu[N(SPiPr2)2]3 to −62.2 in Pu[N(TePiPr2)2]3. Contrastingly, the corresponding diamagnetic La(III) complexes exhibit a 31P NMR shift range from 64.3 ppm for La[N(SPiPr2)2]3 to 29.5 ppm for La[N(TePiPr2)2]3. The largest paramagnetic shifts were observed for the U(III) complexes (5f3 electronic configuration) with values ranging from −525.1 ppm for U[N(SPiPr2)2]3 to −722.6 for U[N(SePiPr2)2]3 (Table 3).
31P NMR shifts (ppm) for M[N(EPR2)2]3 complexes.
| Compound | 31P NMR shift (ppm) |
| U[N(SePPh2)2]3 | −722.6 |
| U[N(TePiPr2)2]3 | −696.7 |
| U[N(SPPh2)2]3 | −680.6 |
| U[N(SePiPr2)2]3 | −573.9 |
| U[N(SPiPr2)2]3 | −525.1 |
| Pu[N(TePiPr2)2]3 | −62.2 |
| Pu[N(SePPh2)2]3 | −59.2 |
| Pu[N(SPPh2)2]3 | −47.1 |
| Pu[N(SePiPr2)2]3 | −20.1 |
| Ce[N(TePiPr2)2]3 | −10.0 |
| Ce[N(SePPh2)2]3 | −8.3 |
| Pu[N(SPiPr2)2]3 | −1.4 |
| Ce[N(SPPh2)2]3 | 4.92 |
| La[N(TePiPr2)2]3 | 29.5 |
| La[N(SePPh2)2]3 | 33.6 |
| La[N(SPPh2)2]3 | 42.6 |
| La[N(SePiPr2)2]3 | 56.8 |
| La[N(SPiPr2)2]3 | 64.3 |
Computational modeling of six-coordinate M[N(EPH2)2]3 complexes, performed by Kaltsoyannis and co-workers at University College London, indicated that enhanced covalency in the M–E bond as the chalcogen group is descended (S to Se to Te) arises predominately from increased metal d-orbital participation, while an increase in f-orbital participation is responsible for an enhancement of covalency in An–E bonds compared to Ln–E bonds [35,36]. Subsequently, calculations on the nine coordinate M[N(EPH2)2]3 complexes demonstrated that N coordination to the metal had little effect on the nature of the M–E bond [37]. Extension of the calculations to Eu, Am, and Cm complexes highlighted the need to experimentally study all of the relevant actinide ions to the extent possible, rather than rely solely on developments in uranium coordination chemistry to drive and refine contemporary theories of actinide electronic structure and bonding [37]. For example, longer than expected Eu–E and Am–E bond lengths were predicted coupled with large f populations. This was interpreted as significant M(II)–E bond character arising from the stabilization of a half full f7 electronic configuration rather than increased covalency in an M(III)–E bond (the radial extension of the f orbitals for M(III) ions in the middle of the series is too small to permit significant metal-ligand orbital overlap). In the case of Cm, the metal ion is very much predicted to behave as Cm(III) and does not exhibit any unusually large f-orbital population. In summary, DFT calculations suggest that f-based covalency is not the origin of any An vs Ln selectivity when considering Am, Cm, Eu complexes. However, the Am and Cm 6d populations are larger than the 5d Eu populations in analogous systems. A computational approach to the ligand system we studied would appear to suggest that the origin of any U vs Ln covalency differences (f-orbital based) could well be different from the origin of Am/Cm vs Ln covalency differences (d-orbital based) with the sandwiched actinides (Np, Pu) exhibiting intermediate behavior. Acquiring experimental data trends across the actinide series (U to Cm) will be hugely beneficial to work in synergy with calculations and aid refinement of the accuracy of computational codes in predicting transuranic behavior.
The X-ray diffraction data for U[N(TePiPr2)2]3 was the first structurally characterized example of a molecular actinide-Te bond, representing a rare case of an extreme, but isolable, ‘very hard’ f-metal – ‘very soft donor’ ligand interaction. As the 5f series is traversed from left to right the An(III) ionic radii decrease and it is expected that the An(III) ions will display even harder Lewis acid behavior, yet interestingly, the analogous Pu–Te could also subsequently be isolated through the synthesis and characterization of Pu[N(TePiPr2)2]3. This finding poses intriguing questions about the importance of hard–soft interactions in f-element chemistry, the extent of metal-ligand valence orbital overlap, and how these properties may be exploited in An/Ln separations for advanced nuclear fuel cycles. In particular, do any covalent properties hold as far across the series as Am(III) and Cm(III)? For Am and Cm, separations involving redox processes are extremely difficult to effect given the high stability of Am(III) and Cm(III). It is hoped that these integrated experimental/theoretical approaches will eventually lead to unequivocal answers about the extent and nature of An(III) vs Ln(III) covalency.
3.4 Pursuit of tetravalent plutonium complexes
In our quest for new Pu(III) organic soluble precursors, we noted that addition of excess iodine or bromine to Pu metal did not lead to an oxidation state higher than Pu(III) [26]. This observation was peculiar to us given that Pu(IV) is the most stable oxidation state under many aqueous solution conditions and that the use of [PuCl6]2− has led to the isolation of a number of Pu(IV) nonaqueous products (section 2). In addition, no oxidation of Pu(III) was observed when [NO][BF4] or TMS-azide were employed as oxidants. An attempt to access the Pu analog of the bis-imido [U(NR)2]2+ moiety through addition of tBuNH2 and iodine to PuI3(py)4 resulted in isolation and structural characterization of [PuI2(thf)4(py)][I3] (Fig. 11) [26]. Interestingly, even dissolution of Pu(IV) carbonate with HCl/Et2O in thf led to partial reduction and isolation of the mixed valent [PuIIICl2(thf)5][PuIVCl5(thf)] complex salt, in low yield, as the only tractable product (Fig. 12) [26]. Given the similar redox chemistry of PuIII/IV and CeIII/IV, it was interesting to note that Lappert et al. have reported that addition of a number of ‘strong’ oxidants to [CeIII(NR2)3] failed to oxidize Ce(III) to Ce(IV). However, addition of TeCl4 did achieve the Ce(III) to Ce(IV) oxidation with formation of [CeIVCl(NR2)3]. Subsequently, our attempts to prepare the Pu analog using this method were successful: Reaction of Pu[N(SiMe3)2]3 with 0.25 equivalents of TeCl4 in toluene afforded the isolation and characterization of PuIV[N(SiMe3)2]3Cl (Fig. 13) [26]. The Pu center is four coordinate with a distorted tetrahedron geometry. This compound has potential synthetic utility to undergo metathesis and deprotonation reactions.
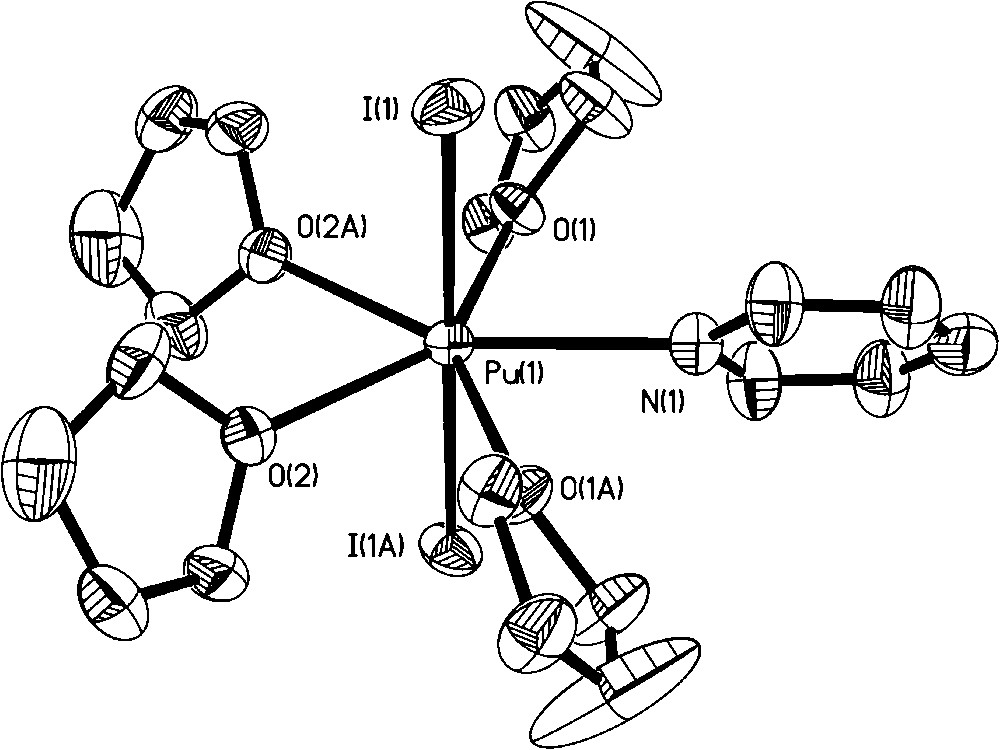
Molecular structure of the [PuI2(thf)4(py)]+ cation. The linear [I3]- anion is omitted for clarity.
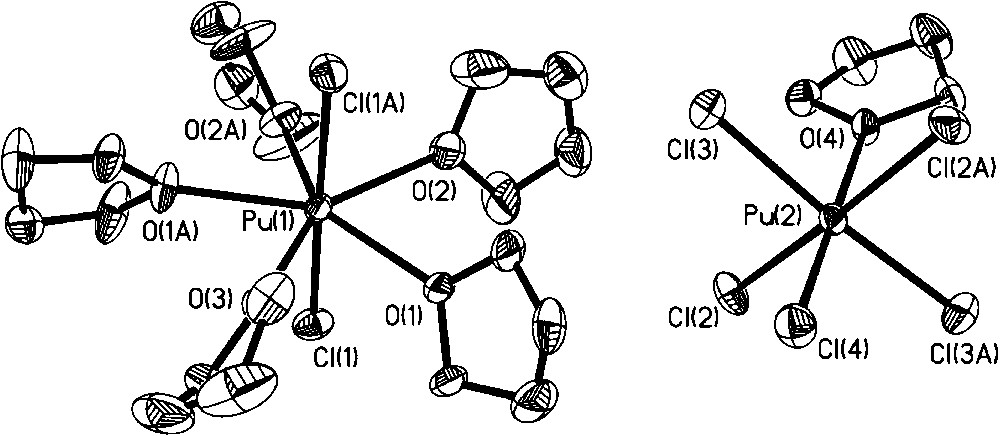
Molecular structure of the [PuCl2(thf)5][PuCl5(thf)] complex salt.
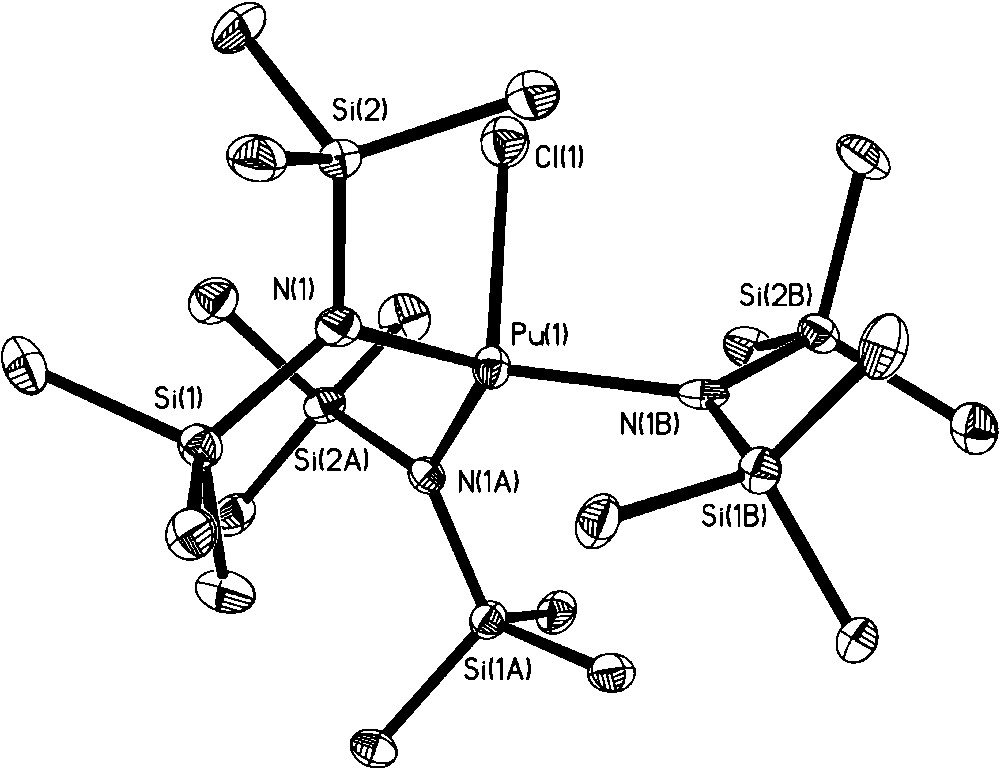
Molecular structure of Pu[N(SiMe3)2]3Cl.
Other Pu(IV) complexes prepared using organic solvents (but not in the absence of water or air) and recently reported in the CSD are Pu(C6H5O3)4 and Pu(C6H4O3Br)4 maltol complexes [38] and Pu(S2CNEt2)4 [25]. In collaboration with the Paine research group (University of New Mexico) we have reported the complexes [Pu(NOPOPO)2(NO3)2][(NO3)2]1.5H2O·0.5MeOH, [Pu(NOPO)2(NO3)2][Pu(NO3)6]0.5, and [Pu(POPO)(OMe)(NO3)3] (NOPOPO, NOPO, and POPO are multidentate ligands comprising aromatic backbones with combinations of pyridine N-oxide and/or phosphine oxide donor functionalities) [39].
3.5 Pentavalent plutonium complexes
We are not aware of any plutonyl(V), PuO2+, coordination chemistry studies yet performed under inert atmosphere nonaqueous conditions. Given the spectacular recent progress in the stabilization of uranyl(V) (see, for example [40], and references therein) and the stability of neptunyl with inorganic ligands, this could be an extremely interesting avenue for future research.
3.6 Hexavalent plutonium complexes
In our publication detailing the preparation and characterization of [(PuO2)2(thf)2Cl2]2 we also described some preliminary investigations of TPPO coordination to the PuO22+ ion [29]. Analysis of the reaction product by UV/vis/NIR spectrophotometry indicates that TPPO did indeed coordinate to PuO22+ in thf solvent. The plutonyl ion with a 5f2 electronic configuration exhibits a characteristic absorption band at 830 nm for the PuO22+(H2O)5 aquo ion, the position of which is highly sensitive to the ligand environment in the equatorial coordination sphere. For example, the absorption band attributed to the PuO22+ species in solution shifts to 838 nm when chloride complexes the ‘aquo-ion’ to form the PuO2Cl+ species. Our preliminary reactivity studies demonstrated that upon addition of TPPO to [PuO2Cl2(thf)2]2 in thf (band at 858 nm), the transition at 858 nm decreases in intensity and a new band at 870 nm appears, a ready indication of TPPO coordination and a PuO22+ speciation change. Solution 31P NMR measurements also indicate that TPPO coordinates to PuO22+ in thf solvent, but the speciation was complex and no crystals suitable for single crystal X-ray diffraction were obtained. However, Russian workers have determined the molecular structure of [PuO2(TPPO)4][ClO4]2 isolated from an acidic Pu residue dissolved in an ethanolic solution of TPPO [41], while a similar procedure involving the addition KNO3 resulted in isolation of [PuO2(NO3)2(TPPO)2] as brown crystals [42].
The current interest in plutonyl chemistry is driven partly by a desire to explore the effect of 5f orbital electronic occupation and actinide contraction across the actinyl series upon reactivity and bonding properties. Again, motivation and a platform for successful ventures is provided by the dynamism of nonaqueous uranyl chemistry over the last several years that has unearthed a number of surprising phenomena and poses intriguing questions about the other actinyl ions [1,9,40,43]. In addition, discovering environments that can stabilize plutonyl and neptunyl species has potential implications for separations application related to nuclear energy and environmental isolation (see, for example [44]). In collaboration with Prof. T.W. Hayton's group (UCSB), we are aiming to probe reactivity and bonding patterns across the actinyl series from uranyl to neptunyl to plutonyl in both the penta- and hexavalent oxidation states.
4 Conclusions and future prospects
We have highlighted the results of our efforts to explore rare nonaqueous plutonium coordination chemistry over the last several years. Through this effort and those of our colleagues, the number of structurally characterized plutonium complexes has grown significantly and in the process enhanced the actinide community's knowledge of reactivity in organic solvents along with data concerning structural preferences and metrical parameters in crystalline compounds. We have developed new Pu(III), Pu(IV), and Pu(VI) precursors to facilitate continued and expanded exploration in this field. Moreover, we have targeted systematic experiments with carefully chosen ligand sets to increase our understanding of bonding trends with soft donors as the f-element series is transgressed. As more research programs of this nature are conducted, from ourselves and others, it is hoped that combining syntheses and structural characterization with modern spectroscopic techniques [45], and increasingly accurate theoretical computational codes, will lead to a much deeper comprehension of the electronic structure and fundamental bonding principles of Pu in a variety of oxidation states, and how they compare to its actinide neighbors and the lanthanides.
The research in this manuscript provides a glimpse of what plutonium coordination chemistry can offer. However, we are only beginning to make substantial progress towards our goal of understanding actinide bonding and translating our findings into practical use. In this respect, we intend to conduct competition/ligand displacement experiments to assess how An/Ln bonding differences with the soft donor ligands affect actinide selectivity and speciation in solution. This line or research will advance our understanding of how the actinide ions engage in covalent interactions and unravel the relative roles of the 6d and 5f orbitals in molecular bonding. We shall also conduct experiments to determine any differences in 1st and 2nd sphere solvation between 4f and 5f series complexes, which can be exploited to separate otherwise similar complexes. Results from these systematic fundamental and applied experiments can address technological needs, such as design of selective chelators for actinide separations in advanced nuclear fuel cycles.
Acknowledgements
We thank the following colleagues for contributing to this work and for engaging in productive collaborations well beyond the scope of this paper: Dr. John Matonic, Dr. Alejandro Enriquez, Mr. Sean Reilly, Prof. Robert Paine, Prof. James Ibers, Dr. Brian Scott, Dr. Evelyn Bond, Prof. Nikolas Kaltsoyannis, Prof. Trevor Hayton, Dr. James Boncella, Dr. Iain May, and Dr. Wolfgang Runde. In particular, Dr. Matonic established laboratory equipment and methods for Pu(III) and Pu(IV) molecular syntheses and characterization, which paved the way for Dr. Enriquez and Dr. Gaunt, who in turn established techniques and chemistry to launch new lines of research. The results reviewed in this account were originally generated through projects funded by the United States Department of Energy (DOE), Office of Science, Basic Energy Sciences, Heavy Element Chemistry Program, the GT Seaborg Institute at Los Alamos National Laboratory, and previous University of California Los Alamos National Laboratory Contractor reinvestment programs. We thank the University of California Office of the President (UCOP) Lab Fees Research Program (collaboration with Prof. Hayton at UCSB), and the DOE, Office of Nuclear Energy, Fuel Cycle Research and Development Program for current transuranic chemistry support. A.J.G. thanks DOE, Office of Science, Early Career Research Program Award for Molecular Transuranic Discovery Science due to commence this year.