

1 Introduction
The choice of ionic liquids (IL) available to synthetic chemists widens every day. New ILs are often obtained by combining a given cation with different anions, so that the properties of the obtained ILs correspond to certain requirements. It is considered necessary that the synthesized ILs can be reused several times and that their degradation products are not toxic [1].
Catalytic reactions make up a significant part of green chemistry processes. ILs show great potential as solvents and catalysts for these processes, due to their unique physicochemical properties. Corma et al. used imidazolium IL with a covalently linked palladium complex for catalysis of Heck and Suzuki–Miyaura cross-coupling. They also demonstrated stability over reuse for this catalyst [2]. Imidazolium ILs have been used as catalysts also in a great variety of other processes, such as hydroformylation, asymmetric hydrogenation, ring-closing metathesis and asymmetric aldol reaction. Catalytic properties of imidazolium ILs have been reviewed recently [3].
Imidazolium-based ILs, particularly chlorides, have also been recognized as perspective biopolymer solvents with ability to dissolve cellulose [4]. Imidazolium ILs bearing alkoxy [5], hydroxyl, and glycidyl groups [6] in the side-chain have been synthesized. Polar substituents in the chain attached to N-1 in the imidazolium ring and chain length determine physicochemical properties of the molecule and at the same time solvent properties of the IL. Relevant information on physicochemical properties of the IL can be obtained by HPLC [7]. This method allows finding the most appropriate conditions for purity determination of the IL and identification of impurities by their sorption parameters.
Recently some ILs have been separated by reversed-phase (RP) [8] and ion exchange (IE) chromatography [9]. The described results of analysis of 1-alkyl- and 1-aryl- 3-methyl imidazolium IL by RP chromatography testify that it is possible to ensure satisfactory indicators of peak tailing and system efficiency by choosing a sorbent with a low content of free silanol groups. Unfortunately, in such a case, poor repeatability of retention factors is observed, which can be improved by stabilizing pH of the mobile phase. In order to be able to maintain the preferred limits of retention factors, it has been suggested to use acetonitrile instead of methanol as mobile phase modifier [10]. Adjustment of separation parameters, keeping the content of mobile phase modifier unchanged, becomes possible by adding salts at different concentrations. However, decreasing the determination limit of the IL 20 times has to be considered a disadvantage [11]. Phosphate buffer solution can be added to the mobile phase to separate hydrophobic 1-alkyl-3-methyl imidazolium IL on RP [12] under the above-mentioned IE conditions. By changing concentration of the buffer solution, it is possible to adjust significantly the retention factor values of the IL. It must be taken into consideration that by increasing acetonitrile content in the buffer solution the elution order of compounds under IE conditions is changing, which is attributed to transition from RP to direct phase sorption mechanism. It should be remarked that it is very difficult to separate hydrophilic cations of IL with relatively short alkyl chains in the described RP and IE systems. Even though such separation has been accomplished by using a polar RP sorbent, adequate column efficiency and asymmetry factor (As) have not been achieved [13]. Peak asymmetry is attributed to the fact that the sorbent surface is heterogeneous and sorption of a low molecular IL mainly occurs by IE mechanism. Due to highly charged points on sorbent surface, sorption and desorption of highly charged sorbates are encumbered more than sorption and desorption of low charged sorbates.
Other studies have concluded that mixed-mode phases allow more variables for fine-tuning separations than are available in classical RP-HPLC. For example, the use of a RP/cation exchange column has been showing good peak shape for ionized bases and significant column efficiency in comparison to analysis on a C18 single-mode column [14].
In this work, we studied performance of a mixed-mode phase (RP 18/Cation 100A) for the determination of sorption parameters of ether-functionalized imidazolium IL. The effect of mobile phase composition and pH of the buffer solution on the separation of IL cations is demonstrated.
2 Experimental
2.1 Equipment
HPLC was performed with a Shimadzu LC20AD series system comprising a diode-array detector (SPDM20A), a vacuum degasser (DGU-20A3), an autosampler (SIL-20A), a thermostatted column compartment (CTO-10ASVP) and data acquisition and analysis were performed by the Shimadzu LC Solution Chromatography Data System Operation. Separations were carried out on a Mixed-Mode RP-C18/Cation 100A column (Alltech; 250 mm × 4.6 mm I.D., 5 μm particle size). The pH of the buffer was measured in aqueous solutions before the addition of acetonitrile. The pH of the solution was measured using a calibrated glass electrode on a Metrohm 780 pH meter at 295.0(±0.10) K. The mobile phases were filtered using a Millipore (Bedford, MA, USA) 0.45 μm filter before use in HPLC. All HPLC experiments were run at 1.0 mL·min−1 and the temperature of 30 °C. The detection wavelength was set at 210 nm. Each value was measured in triplicate and averaged. Five-microlitre sample volumes were injected in each experiment. The system dead time was measured by injecting water into the system. Column was pre-equilibrated with mobile phase before running the actual isocratic experiments.
NMR spectra (1H, 13C) were recorded on a Varian600 instrument and are referenced to residual protons in the solvent (1H), the solvent carbon-13 signal (13C) in CDCl3 at 25 °C.
Infrared (IR) spectra were collected on Avatar 330 FT-IR spectrometer. Samples for IR were dissolved in CHCl3.
Elemental analysis (C, H, N analyzer) of each synthesized IL was performed on Carlo Erba 1108 CHN Elemental Analyser.
The absorption spectra were recorded on Uvikon-930 spectrophotometer by using a 1 cm quartz cell.
2.2 Chemicals and reagents
For preparation of the mobile phase, HPLC grade acetonitrile (Petrochem) was used, as well as deionized water purified using a Milli-Q system (Millipore), NaH2PO4, 85% solution of HPLC grade orthophosphoric acid. The materials used in IL synthesis are as follows: 2-ethoxyethyl chloride (Aldrich 99.0%, redistilled), 2-chloroethyl phenyl ether (Fluka 97.0%, redistilled), 2-chloroethyl vinyl ether (Aldrich 99.0%, redistilled), triethylene glycol monochlorohydrin (Aldrich 96.0%, redistilled), 2-ethoxyethyl chloride (Aldrich 98.0%, redistilled), chloroethanol (Aldrich 98.0%, redistilled), 2-chloroethylmethyl ether (Aldrich 98.0%, redistilled), 1-chloro-2-(2-methoxyethoxy)ethane (Aldrich 98.0%, redistilled), and 1-methylimidazole (Fluka 99.0%, dried over KOH and redistilled).
Structures of analyzed IL and their main properties are given in Table 1.
Structure of room temperature ionic liquids of the series [CnOmmim][Cl].
| Retry | Structure | R | Formula of ionic liquid | Log P [15] |
| 1 | H | [C2OHmim][Cl] | −4.16 | |
| 2 | CH3 | [C3Omim][Cl] | −3.58 | |
| 3 | C6H5 | [C2OBzmim][Cl] | −1.80 | |
| 4 | CHCH2 | [C2OAllymim][Cl] | −2.99 | |
| 5 | CH2CH3 | [C4Omim][Cl] | −3.24 | |
| 6 | (CH2)2OCH3 | [C5O2mim][Cl] | −3.74 | |
| 7 | (CH2)2O(CH2)2OH | [C6O2OHmim][Cl] | −4.49 | |
| 8 | (CH2)2O(CH2)2OCH3 | [C7O3mim][Cl] | −3.91 |
2.3 Preparation of ionic liquids
2.3.1 1-[2-(2-ethoxy)ethyl]-3-methyl imidazolium chloride
2-ethoxyethyl chloride (8.2 mL, 0.075 moL) and 1-methylimidazole (4 mL, 0.050 moL) were stirred for 60 h at 80 °C until two phases formed. The top phase, containing unreacted starting material, was decanted and ethyl acetate (50 mL) was added with thorough mixing. The bottom phase was washed with ethyl acetate (4 × 25 mL), heated at 80 °C and stirred under vacuum (0.5 mmHg) for 2 d. The product [C4Omim][Cl] was obtained as a colourless liquid (9.34 g, 98%). 1H NMR (600 MHz, CDCl3, δ ppm): 10.21 (s, 1H), 7.57 (t, J = 1.7 Hz, 1H), 7.46 (t, J = 1.7 Hz, 1H), 4.42 (t, J = 7.2 Hz, 2H), 3.96 (s, 3H), 3.64 (t, J = 7.2 Hz, 2H), 3.35 (q, J = 10.2 Hz, 2H), 1.0 (t, J = 7 Hz, 3H); 13C NMR (150 MHz, CDCl3, δ ppm): 137.58, 123.16, 123.04, 68.22, 66.62, 49.75, 36.43, 14.86; IR (CHCl3, cm−1): 3381.3, 3140.7, 2954.0, 2857.4, 2359.6, 1571.9, 1456.5, 1377.1, 1294.0, 1172.8; UV spectrum (nm): λmax (H2O) = 211.4; elemental analysis calcd (%) for C8H15N2OCl (190.67): C 50.39, H 7.93, N 14.69; found: C 50.35, H 7.90, N 14.72.
2.3.2 3-methyl-1-(2-phenoxyethyl) imidazolium chloride
2-chloroethyl phenyl ether (10.4 mL, 0.075 moL) and 1-methylimidazole (4 mL, 0.050 moL) were used, and the product [C2OBzmim][Cl] was obtained as a colourless liquid (11.90 g, 99.7%). 1H NMR (600 MHz, CDCl3, δ ppm): 10.48 (s, 1H), 7.66 (t, J = 2.7 Hz, 1H), 7.49 (t, J = 2.7 Hz, 1H), 7.19 (m, 2H, ArH), 6.90 (t, J = 2.4 Hz, 1H, ArH), 6.82 (m, 2H, ArH), 4.81 (t, J = 7.2 Hz, 2H), 4.31 (t, J = 7.2 Hz, 2H), 3.99 (s, 3H); 13C NMR (150 MHz, CDCl3, δ ppm): 157.30, 137.96, 129.53, 123.12, 122.96, 121.68, 114.31, 66.09, 50.37, 36.43; IR (CHCl3, cm−1): 3381.3, 3083.9, 2963.2, 2854.4, 2360.2, 1599.1, 1492.2, 1462.9, 1377.1, 1294.5, 1174.0; UV spectrum (nm): λmax (H2O) = 212.4; elemental analysis calcd. (%) for C12H15N2OCl (238.72): C 60.38, H 6.33, N 11.74; found: C 60.31, H 6.58, N 11.64.
2.3.3 3-methyl-1-[2-(vinyloxy)ethyl] imidazolium chloride
2-chloroethyl vinyl ether (7.6 mL, 0.075 moL) and 1-methylimidazole (4 mL, 0.050 moL) were used, and the product [C2OAllymim][Cl] was obtained as a colourless liquid (9.40 g, 99.7%). 1H NMR (600 MHz, CDCl3, δ ppm): 10.28 (s, 1H), 7.57 (s, 1H), 7.56 (s, 1H), 6.28 (dd, J1 = 14.3 Hz, J2 = 6.6 Hz, 1H), 4.64 (t, J = 3.8 Hz, 2H), 4.11 (dd, J1 = 2.5 Hz, J2 = 14.3 Hz, 1H), 3.98 (m, 6H); 13C NMR (150 MHz, CDCl3, δ ppm): 150.16, 137.60, 122.93, 123.11, 88.10, 65.66, 48.72, 36.34; IR (CHCl3, cm−1): 3381.7, 3144.2, 2959.9, 2359.9, 1573.3, 1460.4, 1377.1, 1285.9, 1169.6; UV spectrum (nm): λmax (H2O) = 211.9; elemental analysis calcd (%) for C8H13N2OCl (188.66): C 50.93, H 6.95, N 14.85; found: C 50.69, H 7.03, N 14.58.
2.3.4 1-{2-[2-(2-hydroxyethoxy)ethoxy]ethyl}-3-methyl imidazolium chloride
Triethylene glycol monochlorohydrin (10.9 mL, 0.075 moL) and 1-methylimidazole (4 mL, 0.050 moL) were used, and the product [C6O2OHmim][Cl] was obtained as a colourless liquid (12.43 g, 99.1%). 1H NMR (600 MHz, CDCl3, δ ppm): 9.75 (s, 1H), 7.72 (t, J = 2.7 Hz, 1H), 7.54 (t, J = 2.7 Hz, 1H), 4.60 (bs, 1H), 4.44 (t, J = 7.2 Hz, 2H), 3.94 (s, 3H), 3.76 (t, J = 7.2 Hz, 2H), 3.60 (t, J = 6.6 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 3.47 (m, 4H); 13C NMR (150 MHz, CDCl3, δ ppm): 137.38, 123.09, 122.99, 72.46, 69.94, 69.67, 68.45, 60.57, 49.18, 36.23; IR (CHCl3, cm−1): 3279.9, 3146.4, 3074.7, 2868.6, 1572.8, 1453.7, 1377.1, 1290.2, 1171.6; UV spectrum (nm): λmax (H2O) = 211.4; elemental analysis calcd (%) for C10H19N2O3Cl (250.73): C 47.91, H 7.64, N 11.17; found: C 47.70, H 7.80, N 11.10.
2.3.5 1-{2-[2-(2-methoxy)ethoxy]ethoxy}ethyl-3-methyl imidazolium chloride
2-[2-(2-methoxyethoxy) ethoxy]ethyl chloride (12.54 g, 0.075 moL) and 1-methylimidazole (4 mL, 0.050 moL) were used, and the product [C7O3mim][Cl] was obtained as a colourless liquid (11.25 g, 85%); 1H NMR (600 MHz, CDCl3, δ ppm): 10.22 (s, 1H), 7.68 (t, J = 2.7 Hz, 1H), 7.49 (t, J = 2.7 Hz, 1H), 4.52 (t, J = 7.2 Hz, 2H), 3.99 (s, 3H), 3.78 (t, J = 7.2 Hz, 2H), 3.56 (m, 6H), 3.45 (t, J = 3.6 Hz, 2H), 3.27 (s, 3H); 13C NMR (150 MHz, CDCl3, δ ppm): 137.57, 123.37, 122.72, 71.58, 70.08, 70.01, 70.01, 68.83, 59.63, 49.35, 36.23; IR (CHCl3, cm−1): 3389.1, 3146.9, 3077.0, 2901.3, 1572.9, 1456.4, 1377.1, 1290.3, 1171.6; UV spectrum (nm): λmax (H2O) = 211.8; elemental analysis calcd (%) for C11H21N2O3Cl (264.75): C 49.90, H 8.00, N 10.58; found: C 49.73, H 8.05, N 10.48.
The products [C2OHmim][Cl], [C3Omim][Cl] and [C5O2mim][Cl] are known compounds and spectral data are in accordance with the corresponding literature values [16].
3 Results and discussion
The Alltech mixed-mode RP-C18/cation exchange column that we used for separation of ILs has C18 groups and carboxyl groups. Depending on elution conditions and properties of the solutes used, it offers multimode retention mechanisms, including RP, cation exchange, and hydrophilic interaction. These interactions can be controlled by changing pH of the mobile phase. In acidic medium carboxylic, groups of the sorbent are not ionized and retention occurs mainly by the RP mechanism. Then, compound retention is governed by hydrophobic interactions, which can be weakened by increasing the concentration of the organic component in the mobile phase. In basic medium carboxylic, groups of the sorbent are ionized, and both cation exchange and hydrophobic interaction contribute to retention. In highly organic mobile phase, the mixed-mode column can serve as a hydrophilic interaction liquid chromatography (HILIC) column.
The retention behaviour of imidazolium IL was studied by varying the percentage of acetonitrile in the mobile phase at constant sodium dihydrogen phosphate concentration (0.01 M) and pH 6.5. In Fig. 1, the retention factor of the imidazolium IL is plotted as a function of acetonitrile content in the mobile phase. As shown in Fig. 1, the k-φMeCN plot exhibits three different series of curves.
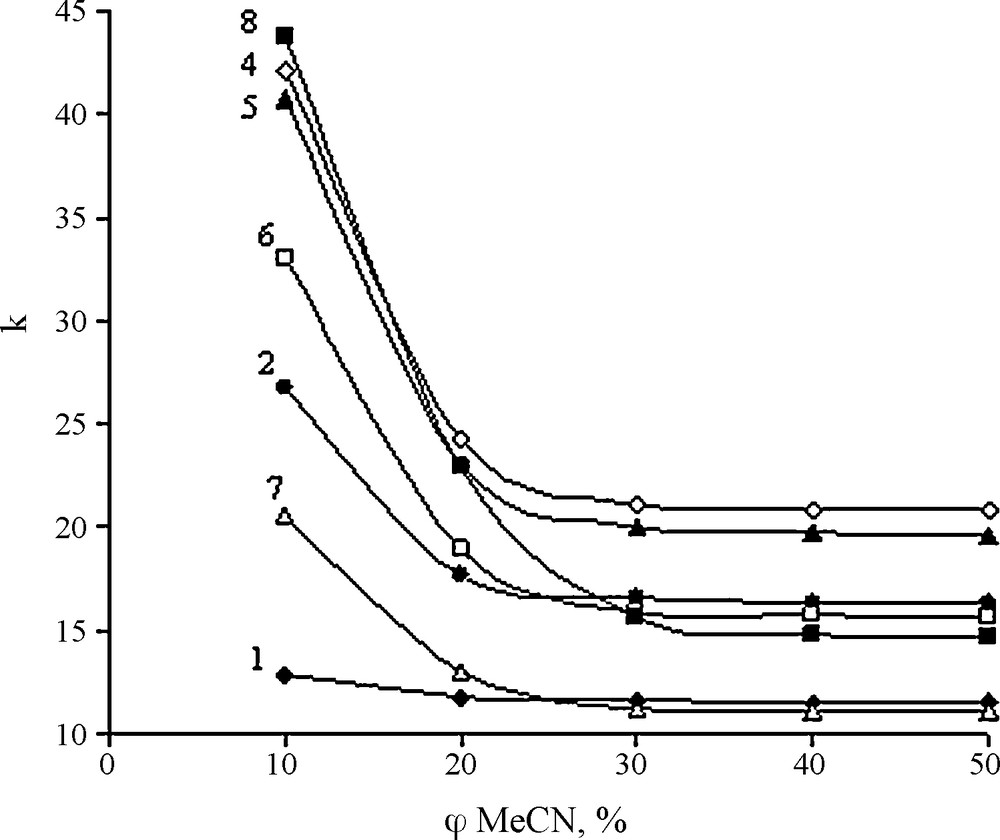
Influence of acetonitrile in mobile phase on retention factors (k) for 1-alkyl ether-3-methylimidazolium ionic liquid (pH = 6.5). The chemical structure (Table 1).
ILs usually possess a strongly polar (charged) group along with a strongly hydrophobic alkyl group. However, this is not the case. Ether groups in IL 1-8 are not as hydrophobic as simple alkyl groups. These groups are capable of interacting not only with a non-polar sorbent, but also with a polar mobile phase. So, there is dynamic equilibrium that is easily shifted by changing mobile phase composition. It is evident that polyether groups are capable of stronger interaction with the sorbent at low acetonitrile content. It is most prominent for compound 8 that has the longest polyether chain. At low acetonitrile content, compounds 4 and 5 that contain ether groups with fewer carbon atoms are eluted just before compound 8. Compounds 6 and 2 that contain substituents terminating in the shortest ether group and the rather hydrophilic compounds 1 and 7 have even lower retention times. Increasing of acetonitrile content results in reduction of retention times for all solutes, but also changes order of elution for several IL. Most markedly retention time is decreased for compound 8, for which hydrophobic interaction at low acetonitrile content in the mobile phase may be considered most significant. In this acetonitrile concentration range and pH value, the retention factor of compound 3 was not obtained (k > 50) because it was not eluted from the column.
When the mobile phase contains more than 30% acetonitrile, the ILs are eluted in the order of increasing lipophilicity coefficients (log P) (Table 1). Log P values were calculated using Marvin and Calculator Plugin Demo [15]. This software uses a sum of group contributions model that has been compiled based on experimentally determined log P values of different molecules. These results demonstrate that all the ILs follow the reverse phase mechanism of sorption and their retention factors remain constant at acetonitrile content above 30%. If acetonitrile content is under 30%, elution of imidazolium ILs does no longer occurs in the order of increasing lipophilicity factors. Obviously, this model is too simplified for complex molecules.
A hydroxyl group in the side-chain of imidazolium ring in compounds 1 and 7 lowers retention more than one or two ethoxy groups at an acetonitrile content of both 50% and 10%. The retention-lowering effect of the hydroxyl group is probably caused by direct hydrogen bond formation to the eluent constituents. It is necessary to note that change of sorption character of alkyl imidazolium IL was also observed in an earlier study in which a C18 single-mode column was used, but this effect was not marked using IE column [9].
The pH of the mobile phase containing 0.01 M sodium dihydrogen phosphate in acetonitrile-water was varied by adding sodium hydroxide or phosphoric acid. In Fig. 2 the k values of imidazolium IL are presented as a function of eluent pH. Fig. 2 shows that k does not increase up to pH 3–4. It can be presumed that at low pH carboxylic groups in the stationary phase remain protonated, and their interaction with positively charged imidazolium rings is weak. Large increase in retention for IL was observed in pH range from 4.0 to 6.5. In this case, it is likely that IL are retained due to IE. Mobile phase pH seems to exert a greater influence on k values of the more lipophilic IL, because the curve corresponding to compound 3 is the steepest.
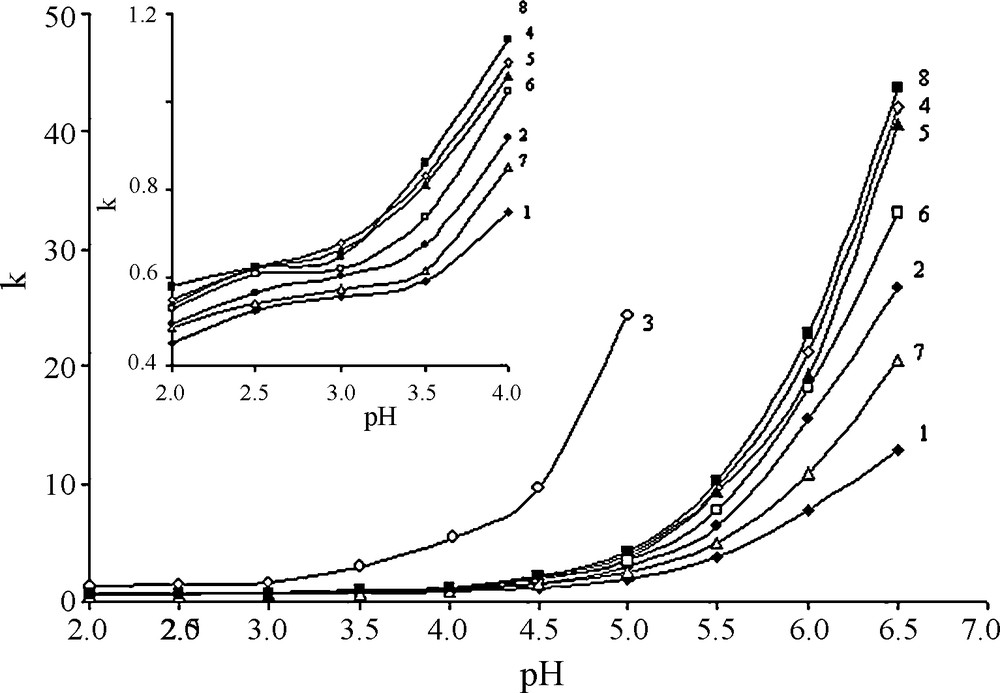
Effect of pH of the mobile phase on retention factor k (φMeCN = 10%). The chemical structure (Table 1).
Efficiency of the C18/cation exchange column was studied and characterized by the number of theoretical plates (N) as a function of mobile phase composition and pH under otherwise identical chromatographic conditions. The asymmetry factor (As) was calculated as the ratio of widths of the rear and front sides of the peak at 10% peak height.
In Fig. 3.A, N and As are plotted as functions of acetonitrile content in the mobile phase. In general, addition of acetonitrile provides more efficient analysis (higher number of theoretical plates N), particularly within φMeCN range of 30–50%. The most significant increase in N is observed for acetonitrile content 10–30%, if the pH is 5. As N increases, As values for the IL decrease.
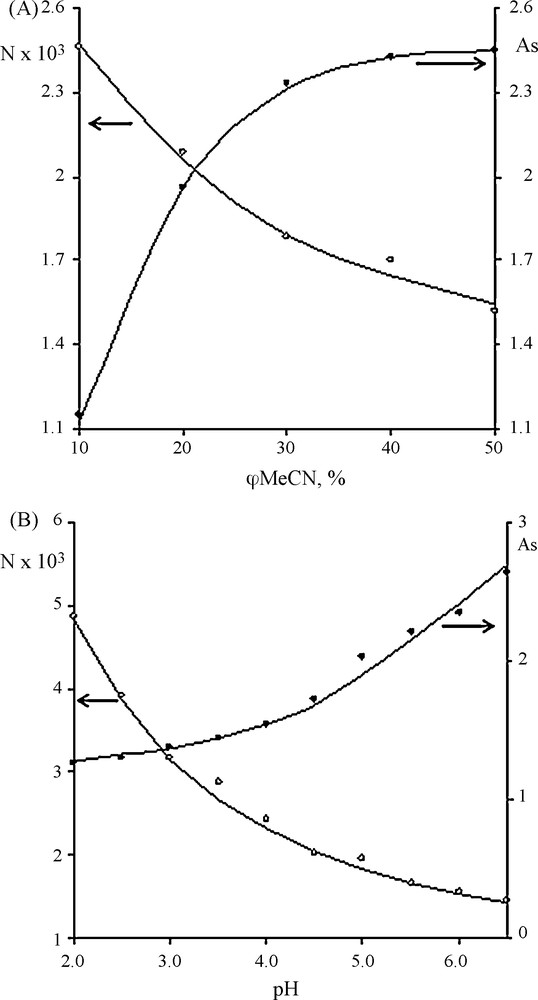
Dependence of the number of theoretical plates N and asymmetry factor As on acetonitrile content in the mobile phase (pH = 5.0) (A) and on pH of buffer solution (φMeCN = 20%) (B) of 1-{2-[2-(2-methoxy)ethoxy]ethoxy}ethyl-3-methyl imidazolium cation.
The relationship observed between pH of the mobile phase and peak asymmetry factor or number of theoretical plates is displayed in Fig. 3.B. At higher pH N decreases and peak tailing is observed. The lowest As can be achieved by use of acidic eluents.
The selectivity of systems is demonstrated in separation of eight imidazolium IL, as shown in Fig. 4. Selectivity (α) and resolution (Rs) values for the IL under isocratic and gradient conditions are shown in Table 2. Under isocratic conditions satisfactory separation was not achieved (Fig. 4A), and gradient elution was preferred. By using acetonitrile as the organic modifier on mixed-mode stationary phase, partial separation of all analyzed IL was achieved by gradient elution (Fig. 4B). Compounds 2 and 3 could not be completely separated.
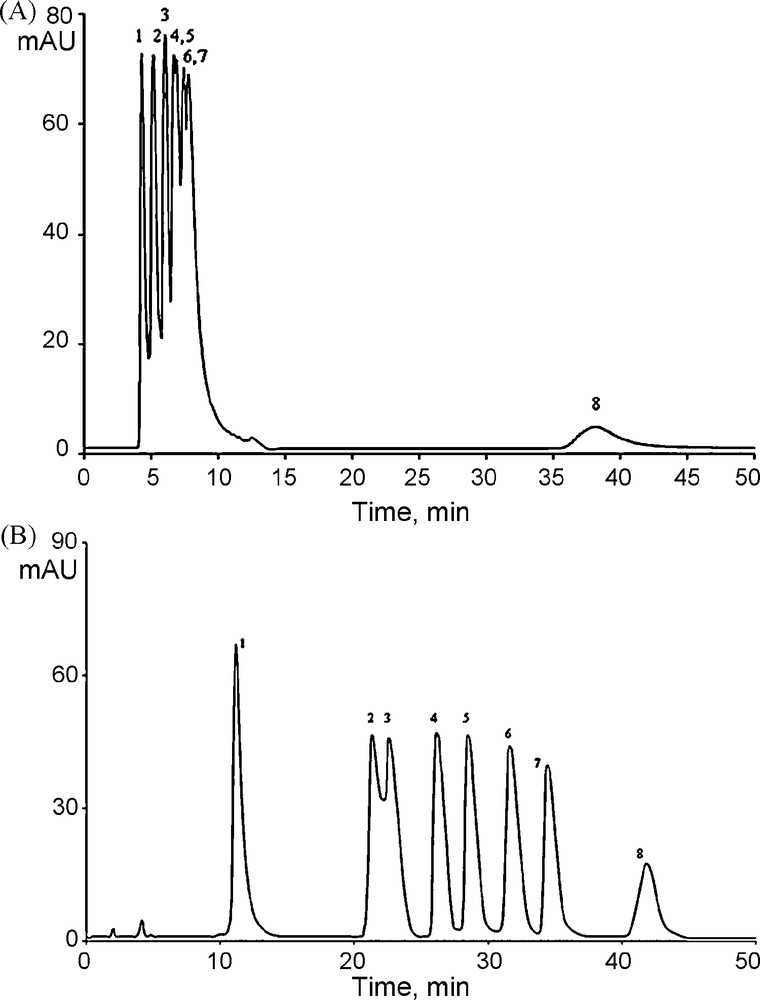
Chromatograms of separation of ether-functionalized imidazolium ionic liquids. A. Isocratic elution, mobile phase 10% CH3CN/10 mM NaH2PO4/H3PO4, pH 5.0. B. Gradient elution. The concentration ratio of acetonitrile to buffer solution at the gradient changed: 0 min, 0%, CH3CN; 3–6 min, 2% CH3CN; 12–20 min, 5% CH3CN; 20–40 min, 10% CH3CN. Column: Alltech Mixed-Mode RP-C18/Cation 100A column (250 mm × 4.6 mm I.D., 5 μm). Sample concentration in mobile phase: 0.5 mg·mL−1. 1 – [C2OHmim][Cl], 2 – [C6O2OHmim][Cl], 3 – [C3Omim][Cl], 4 – [C5O2mim][Cl], 5 – [C4Omim][Cl], 6 – [C2OAllymim][Cl], 7 – [C7O3mim][Cl], 8 – [C2OBzmim][Cl].
Separation selectivity (α) and resolution (Rs) (the chromatographic conditions were the same as in Fig. 4).
| Ionic liquid | Isocratic | Gradient | ||
| α | Rs | α | Rs | |
| [C2OHmim][Cl] | ||||
| 1.32 | 0.56 | 2.05 | 4.52 | |
| [C6O2OHmim][Cl] | ||||
| 1.24 | 0.50 | 1.06 | 0.52 | |
| [C3Omim][Cl] | ||||
| 1.13 | 0.34 | 1.16 | 1.65 | |
| [C5C2mim][Cl] | ||||
| 1.02 | 0.05 | 1.10 | 1.32 | |
| [C4Omim][Cl] | ||||
| 1.13 | 0.32 | 1.12 | 1.49 | |
| [C2OAllymim][Cl] | ||||
| 1.05 | 0.12 | 1.09 | 1.25 | |
| [C7O3mim][Cl] | ||||
| 5.75 | 8.01 | 1.22 | 3.13 | |
| [C2OBzmim][Cl] |
As mentioned before, at high acetonitrile content and in an acidic medium k becomes smaller, and so does the difference between the analyzed IL. This makes use of acidic eluents with a large percentage of acetonitrile impractical for separation of the analyzed IL. So, the choice of proper chromatographic conditions presents a compromise between resolution, reasonable retention times and acceptable peak shape. Taking into account all chromatographic properties of the investigated compounds (α, Rs, k, N, T) under discussed conditions, it can be seen that the best chromatographic results can be obtained by use of systems with pH = 5.0–6.0 and φMeCN = 10–30%.
4 Conclusions
A complementary set of ether-functionalized imidazolium IL have been successfully prepared from 1-methylimidazole and the corresponding chlorides. An HPLC method for separation and determination of ILs using a RP/cation exchange type column has been developed. This column has given strong retention of IL cations at pH close to neutral. The IL eluted from the column in order of increasing lipophilicity factor, if the contents of acetonitrile in the eluent was higher than 30%. Partial separation of the studied IL has been achieved under gradient conditions using a buffered eluent that contained acetonitrile.