

1 Introduction
Molten salts with melting points below 100 °C are commonly denoted ionic liquids (ILs) and have, during the last years, received a tremendous amount of attention owing to their usability for a variety of applications [1]. ILs are nowadays employed in battery electrolytes, separation processes, as lubricants and – not only at a lab scale – as (catalytically active) solvents in synthetic chemistry [2,3]. The chemical nature of ILs was during the last two decades dominated by the combination of stable tetraalkylammonium (I, Chart 1), 1,3-dialkylimidazolium (II) or N-alkyl-pyridinium (III) cations with suitable anions such as halides, complex halides, triflate, or triflamide [1]. Compared to the use of the nitrogen-based cations, phosphorus-containing species have much less frequently been employed for the design of ILs, although some specimen containing tetrasubstituted phosphonium cations (IV, R1,2 = alkyl/aryl) are known [4–7] and have also proven useful for selected applications [7].
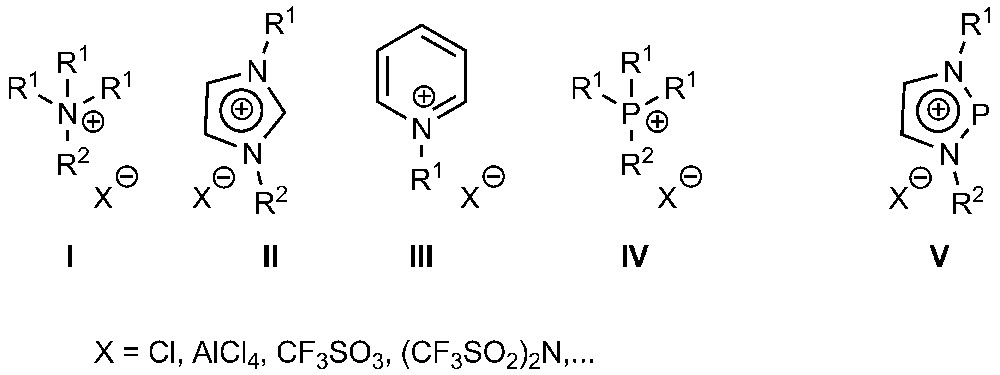
Important types of ionic liquids.
A well-known aspect of phosphorus chemistry is that beside phosphonium ions [R4P]+ a second class of stable cations exists. These phosphenium ions of composition [R2P]+ are distinguished by a divalent rather than a tetravalent central atom and can generally be isolated in the form of thermally stable salts if the electron deficiency at the phosphorus atom is intramolecularly mediated by suitable π-donor substituents (in most cases amino groups) [8]. A particularly interesting group of such cations are N-heterocyclic diazaphospholium ions V, which were first synthesized by the groups of Pudovik [9], Denk [10], and Cowley [11], and can be regarded as isovalent analogues of 1,3-disubstituted imidazolium ions II. These cations exhibit both an exceptionally high stability [12] and a peculiar reactivity, e.g., owing to their ability to bind as ligands to transition metals [13,14], or to act as Lewis acidic catalysts [15]. Although these features would make their use in ILs rewarding, it appears that, as yet, no such attempts have been undertaken. In view of the fact that in imidazolium-based ILs the presence of different substituents on the heterocyclic core of the cations has generally a beneficial effect on the desired melting behavior, the best candidates for phosphenium-based ILs would be cations V with an unsymmetrical substitution pattern (i.e., R1 ≠ R2). Since the reported syntheses [9–11,16] focused mainly on symmetrical derivatives (with R1 = R2), we felt the need for the development of a dedicated synthetic route to unsymmetrically substituted N-heteroyclic phosphenium ions V. We report here on two approaches which allow the assembly of monocyclic or benzannulated derivatives with different N-substituents, and the characterization of the triflates by spectroscopic, thermoanalytical measurements and, in selected cases, by single-crystal X-ray diffraction studies.
2 Results and discussion
The ring system of N-heterocyclic phosphenium ions V is commonly assembled by joining a P1 fragment with a NCCN building block which is normally derived from an α-diimine precursor [9–11,16]. The synthesis of derivatives with two different N-substituents according to the same route would then require the use of a non-symmetrically substituted diimine precursor. Compounds of this type are accessible according to Rieger et al. [17] who performed first a condensation of a diketone with an equimolar amount of a bulky primary amine to give an isolable ketimine, and then coupled this product with a second amino component. Following this approach, we prepared the non-symmetrical aryl/alkyl diimine 1 by stepwise reaction of diacetyl with 2,6-diisopropylaniline and cyclohexylamine. Conversion of this product into the non-symmetrical P-chloro-diazaphospholene 2 and subsequent chloride abstraction with trimethylsilyl trifluoromethanesulfonate (triflate) to give the salt 3 was then accomplished by using the same protocol as had been employed for the synthesis of symmetrical diazaphospholenes (Scheme 1) [16].
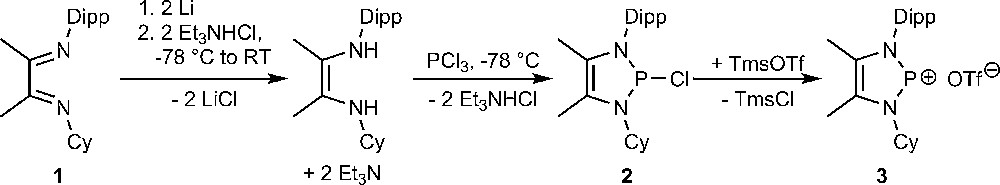
Synthesis of a N-alkyl-N′-aryl-1,3,2-diazaphospholium salt (Dipp = 2,6-diisopropylphenyl, Cy = cyclohexyl, Tms = Me3Si, OTf− = triflate).
Both phosphorus heterocycles 2 and 3 were characterized by analytical and spectroscopic data. The 1H and 13C shifts are as expected, and the 31P chemical shifts match those of known symmetrically substituted derivatives [18]. The P-chloro-diazaphospholene 2 was further characterized by a single-crystal X-ray diffraction study. The asymmetric unit contains two crystallographically independent molecules whose structural parameters (Fig. 1) are very similar to each other and also to those of the symmetrical 2-chloro-1,3-bis(2,6-diisopropyl)phenyl-4,5-dimethyl-1,3,2-diazaphospholene (4, Chart 2). Worth mentioning is the finding that the P–Cl bond (2.357(2)/2.340(2) Å) in 2 is slightly but perceptibly longer than in 4 (2.325(1) Å) [18], which reflects not only the commonly observed ionic bond polarization in P-chloro-diazaphospholenes, but fits also into the trend that this effect becomes particularly large for compounds with N-alkyl substituents, presumably as a consequence of the superior σ-donor capability of N-alkyl as compared to N-aryl groups [18]. Analysis of the thermal behavior of 3 by means of differential scanning calorimetry (DSC) disclosed that around 32 °C a glassy phase was formed which displayed no further detectable phase transitions until above 310 °C an irreversible decomposition was observed.
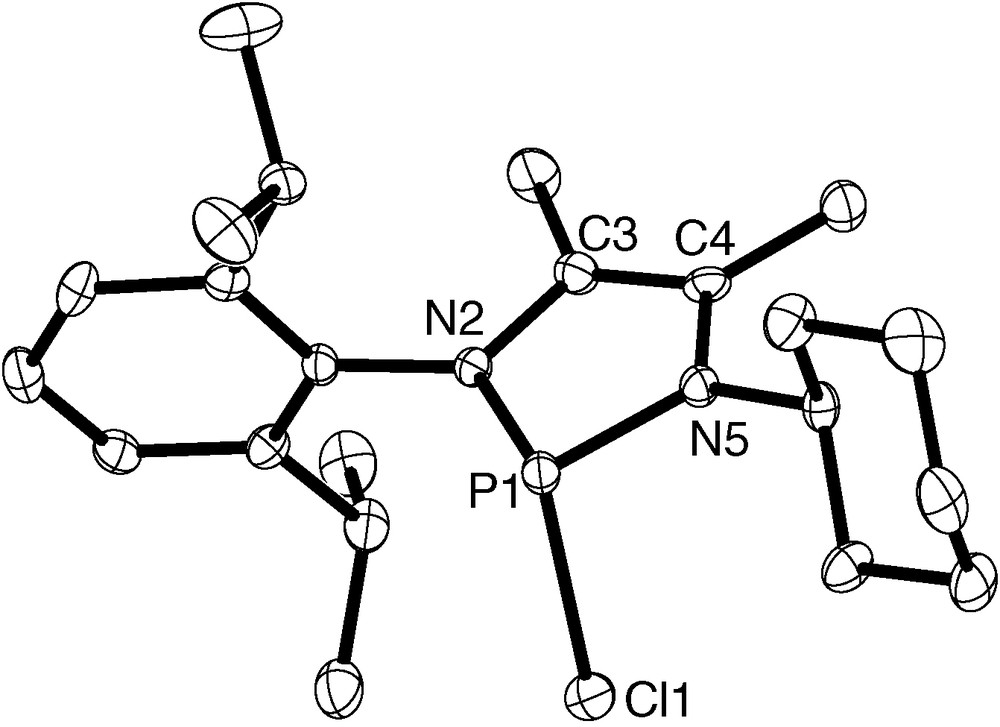
Molecular structure of one of the crystallographically independent molecules in crystalline 2 (H–atoms omitted for clarity; 50% probability thermal ellipsoids); selected bond lengths [Å] (values for the second molecule are given in brackets): Cl1–P1 2.357(2) [2.340(2)], P1–N5 1.669(4) [1.663(4)], P1–N2 1.674(4) [1.672(4)], N2–C3 1.421(7) [1.424(6)], C3–C4 1.358(7) [1.354(7)], C4–N5 1.412(6) [1.419(6)].

Molecular structure of 4 (Dipp = 2,6-diisopropylphenyl).
Even though the synthetic approach leading to 3 should allow to access a variety of mixed aryl/alkyl-substituted diazaphospholene derivatives, these are limited to sterically hindered products with at least one extremely bulky N-aryl substituent.1 In order to circumvent this restriction, we considered an alternative scheme which should also permit the preparation of non-symmetric dialkylated N-heterocyclic phosphenium ions without steric protection. The basis of our approach are earlier results by Malavaud et al. [19] who established that N-alkyl-benzo-1,3,2-diazaphosphole tetramers 5 (Scheme 2) react with Lewis acids under cleavage of the tetramer and formation of monomeric Lewis complexes, and by Lopez et al. [20] who used the reaction of 5 with acyl halides to produce asymmetrically substituted N-acyl-N′-alkyl-2-chloro-benzodiazaphospholenes. As both reactions proceed with electrophile induced cleavage of the tetramers and attachment of an electrophile to the unsubstituted nitrogen atom, we reckoned that a similar electrophilic alkylation of 5 should render the desired N,N′-dialkyl-benzodiazaphospholene derivatives. In line with this anticipation, we found that 5a–d reacted with methyl triflate (“magic methyl”) to give in one step the N-alkylated benzo-1,3,2-diazaphospholium triflates 6a–f (Scheme 2) which were isolated in yields of 60–92% after removal of the solvent. A similar reaction as with methyl triflate was also observable between 5 and triflic acid and enabled the formation of NH-substituted phosphenium salts. As a prominent representative of such compounds, we isolated the salt 8 with the parent benzo-1,3,2-diazaphospholium ion [21]. This species constitutes one of still very few examples where stabilization of a phosphenium cation relies exclusively on π-delocalisation without contribution by steric or inductive influences of N-substituents,2 and exhibits Brønsted acid properties [21]. It should be noted that a tetrachloroaluminate salt containing the same symmetric cation as 6a had previously been obtained via Lewis-acid promoted halide abstraction from a P-chloro-substituted precursor [23].
The salts 6a–f and 8 were characterized by analytical and spectroscopic studies and, in case of 6d, f, also by single-crystal X-ray diffraction studies. The NMR data of all compounds do not deviate significantly from one another and match the previously reported data of 1,3-dimethyl-benzo-1,3,2-diazaphospholium tetrachloroaluminate [23] and monocyclic 1,3,2-diazaphospholium triflates [18]. The individual bond distances and angles as well as the shortest intermolecular contacts in 6d, f (Figs. 2 and 3) are likewise closely similar to those of 8 [21] and previously published data of other N-heterocyclic phosphenium salts [18,23], but the two structures show nonetheless, some interesting differences in molecular conformations and crystal packing. Thus, the cations in crystalline 6d are arranged in a way that the planar heterocyclic ring systems form shifted π-stacks along the crystallographic a-axis (Fig. 2). The center of gravity of adjacent benzo-diazaphospholenium units in each stack is horizontally displaced from the common axis, and the phosphorus atoms point into opposite directions. The n-butyl chains display a gauche-conformation at the α-carbon atom, and are thus rotated out of the aromatic ring plane and oriented in such a way as to group adjacent cations in the stack into centrosymmetric pairs. The triflate anions are arranged in piles that fill the gaps between the cation stacks. Two oxygen atoms of each anion exhibit intermolecular contacts to the phosphorus atoms of every other unit in the cation stack (Fig. 2; the distances of 3.11 Å (P1–O1) and 3.09 Å (P1–O2) are of similar magnitude as in other N-heterocyclic phosphenium triflates [18,21]), so that the whole crystal structure can be described as a columnar array of interlocking one-dimensional chains of mutually connected cations and anions.
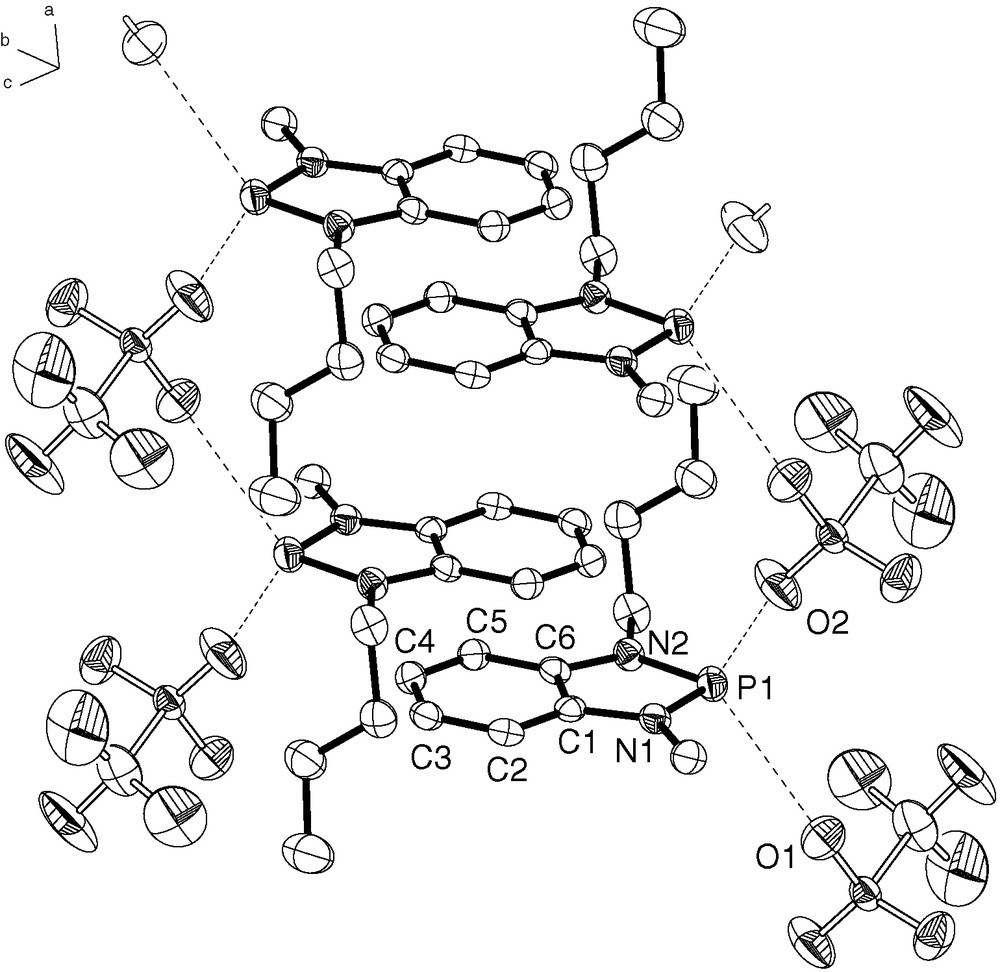
Section of one of the columnar stacks showing the packing of anions and cations in crystalline 6d (H-atoms omitted for clarity; 50% probability thermal ellipsoids); selected bond lengths [Å]: P1–N1 1.649(2), P1–N2 1.665(2), N1–C1 1.396(3), N2–C6 1.392(3), C6–C1 1.399(3), C6–C5 1.404(3), C5–C4 1.376(3), C3–C2 1.382(3), C3–C4 1.406(3), C2–C1 1.394(3), intermolecular distances [Å]: P1–O1 3.111(2), P1–O2 3.092(2) (O1 and O2 are symmetry generated; operators: O1: −x, 1 − y, 1 − z; O2: 1 − x, 1 − y, 1 − z).
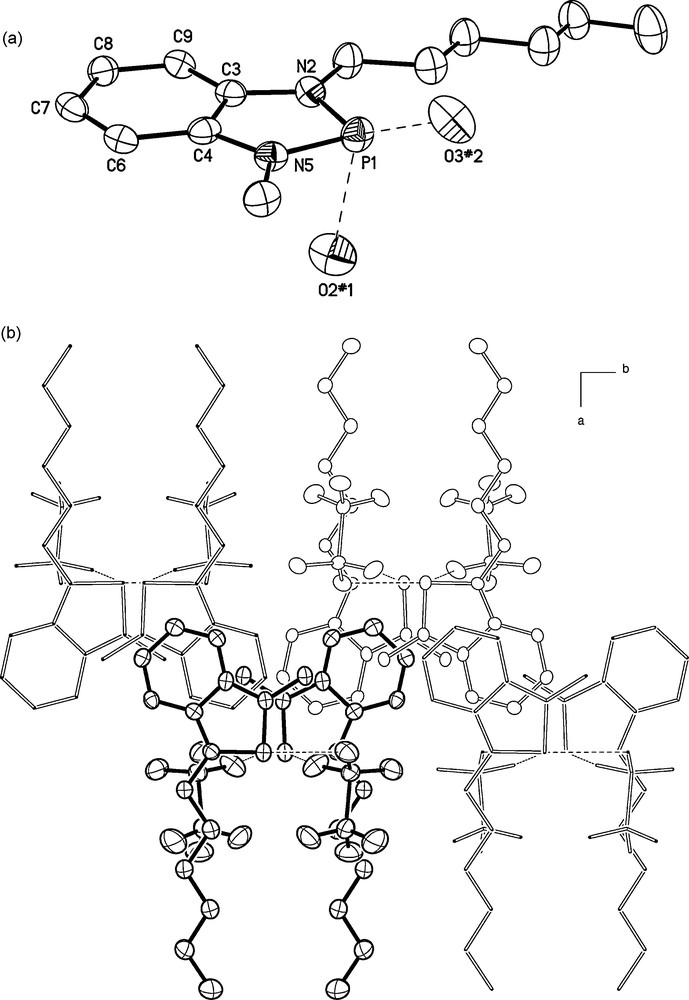
(a) Molecular structure of a single cation in crystalline 6f showing the atom numbering scheme and the closest intermolecular cation–anion contacts (H–atoms omitted for clarity; 50% probability thermal ellipsoids); selected bond lengths [Å]: P1–N5 1.648(3), P1–N2 1.658(3), N2–C3 1.385(5), N5–C4 1.397(4), C3–C4 1.400(5), C4–C6 1.396(5), C6–C7 1.374(5), C7–C8 1.413(5), C8–C9 1.369(5), C9–C3 1.401(5), intermolecular distances [Å]: P1–O2#1 3.047(3), P1–O3#2 3.126(3) (#1: −x + 1, −y + 2, −z + 1; #2: −x + 1, y − 0.5, −z + 0.5). (b) Representation of a part of the crystal packing of 6f viewed along the crystallographic c-axis showing sections of four of the columnar cation-anion stacks linked by PO contacts.
In contrast, the n-hexyl chains of the cations in crystalline 6f display an all-trans conformation and are practically aligned in the plane of the fused aromatic ring system (Fig. 3). The phosphorus atom of each cation exhibits close intermolecular contacts to oxygen atoms of two triflate ions (P1–O2 3.05 Å, P1–O3 3.13 Å) which leads, in a similar way as in 6d, to the formation of zigzag chains of alternating anions and cations in the direction of the crystallographic c-axis (Fig. 3). Each cation exhibits further a π-stacking interaction to a cation in a neighboring chain which links parallel one-dimensional ion stacks together to give a two-dimensional double-layer superstructure which is terminated on both sides by n-hexyl substituents and is oriented parallel to the crystallographic b,c-plane (Fig. 3). The third dimension of the crystal is generated by stacking the double-layers with an interlocking arrangement of the hexyl-substituents in the cations, and a pairwise grouping of the CF3-groups in the anions, of adjacent layers. The whole arrangement has a certain appeal as the individual cations and anions in the crystal interact in one direction by (electrostatically dominated) PO contacts, in the second direction by π-stacking interactions, and in the third dimension preferably by dispersive interactions between (fluoro)alkyl groups.
A study of the thermal behavior of 6a-f by differential scanning calorimetry (DSC) revealed that all compounds melt reversibly between 80 and 125 °C and are thermally stable up to at least 200 °C. A plot of fusion temperatures vs. the chain length of the N-alkyl group R1 (Fig. 4) shows that derivatives with even-numbered chain lengths of R1 melt at slightly lower temperatures than those with odd-numbered chain lengths (just opposite to the known behavior of n-alkanes), and that the lowest fusion points of all compounds are observed for the N-ethyl and N-butyl derivative, respectively. All DSC curves give evidence for at least one exothermal process during the cooling phase which we interpret as solidification; the associated temperatures show no alternating behavior but decrease continuously with growing chain length of R1. In case of 6b, recording the DSC curve through repeated heating/cooling cycles revealed that regular solidification during the cooling phase was only observed for the first cycle, whereas in the remaining cycles delayed solidification occurred only during the warm-up phase. A similar behavior which indicates a strong tendency to form supercooled liquids is also known for imidazolium-based ILs [24]. The DSC curves of the lighter homologues 6a–c display further a second exothermic peak at lower temperature (with energies amounting to 25–50% of that of the first exothermic process), which we assign to a further phase transition. The observed behavior during a full heating/cooling cycle suggests that this transition is not reversible, and that the intermediate phase is upon re-heating only accessible via the melt. Whereas the energies of all endothermic and exothermic processes match well for 6a–d, the fusion energies of 6e,f are by some 17–25% higher than the energies of the endothermic process; furthermore, visual inspection of samples of crystalline 6e,f that had been subjected to a melting/cooling cycle in a melting-point determination apparatus revealed a glassy rather than a specific crystalline appearance. We interpret these findings by assuming that cooling of molten samples of 6a–f produces first a phase that is thermodynamically less stable than the starting material and is possibly amorphous. The observation of a second exothermic phase transition for 6a–c is then attributable to conversion of the unordered into a crystalline phase at lower temperature, whereas in case of 6d both processes seem to occur at comparable temperatures and are therefore not resolved. In case of 6e,f, where the first phase transition occurs at still lower temperature, crystallization is apparently totally inhibited, presumably as a consequence of a significantly increased viscosity.
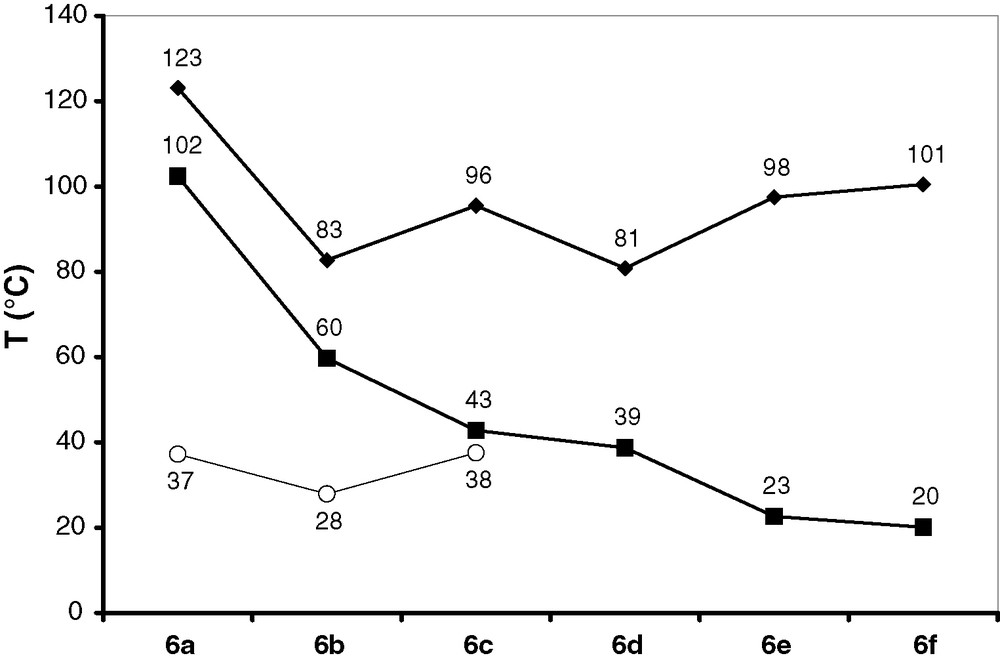
Melting temperatures (diamonds) and solidification temperatures (squares) of 6a–f. Circles denote the temperature of a second exothermal process during the cooling phase which we assign to a further phase transition.
3 Conclusions
Two approaches for the controlled synthesis of 1,3,2-diazaphospholium cations with two different N-substituents have been derived. Reduction and condensation with PCl3 of unsymmetrically substituted α-diimines should give access to both unsymmetrical aryl/alkyl and diaryl cations with isolated rings, but is restricted to the preparation of derivatives with at least one sterically demanding substituent. In contrast, electrophilic depolymerisation and alkylation/protonation of benzo-1,3,2-diazaphosphole-tetramers provides unsymmetrically substituted benzannulated N-heterocyclic phosphenium ions which need not be stabilized by bulky substituents. Single-crystal X-ray diffraction studies of two of these phosphenium salts give insight into interesting supramolecular interactions in the crystalline state. Studies of the thermal behavior of benzodiazaphospholium triflates reveal that melting temperatures are higher than those of common imidazolium-based ILs, but that, in particular, cations with medium length alkyl chains melt below 100 °C and display a strong tendency to form supercooled liquids at even lower temperatures. These results should encourage further investigations of the use of these phosphenium salts as ionic liquids.
4 Experimental section
All manipulations were carried out under an atmosphere of dry argon using standard vacuum line techniques. Solvents were dried by standard procedures. Cyclotetra-phosphazanes were prepared as previously described by Malavaud et al. [19]. 2,3-Butanedione-(2,6-diisopropylphenyl)imine was synthesized as described by Rieger et al. [17]. NMR spectra were recorded on Bruker Avance 400 (1H: 400.1 MHz, 13C: 100.5 MHz, 31P: 161.9 MHz) or Avance 250 (1H: 250.1 MHz, 13C: 62.8 MHz, 31P: 101.2 MHz) NMR spectrometers at 303 K. Chemical shifts are referenced to ext. TMS (1H, 13C) or 85% H3PO4 (Ξ = 40.480747 MHz, 31P), and coupling constants are given as absolute values if not indicated otherwise; i, o, m, p denote the positions in N-aryl rings. EI-MS: Varian MAT 711, 70 eV. Elemental analysis: Perkin-Elmer 24000CHN/O Analyzer. Differential scanning calorimetry (DSC) measurements were performed with a NETZSCH DSC-204 Phoenix thermal analyzer using heating and cooling rates of 10 K min−1 (in some experiments also rates of 5 K min−1 were used), respectively. Sample preparation was carried out under inert atmosphere in a glove box. The given fusion and solidification temperatures represent the onset points of the exothermic or endothermic peaks in the DSC curves which are independent on heating or cooling rates, respectively. Molar heats of fusion could not be determined due to the lack of a possibility to determine sufficiently accurate sample weights under inert atmosphere conditions. Evaluation of the areas under all peaks observed during one heating/cooling cycle indicated that for 6a–d, the energies of the exothermic and endothermic processes are comparable (deviations of <10% are presumably to a good deal attributable to the temperature dependence of the heat capacities); for 6e,f, the fusion energy is by 17–25% higher than the energy of the exothermic phase transition observed during the cooling phase.
1-cyclohexyl-4-(2,6-diisopropylphenyl)-1,4-diazabutadiene 1: A flask containing a solution of 2,3-butanedione-(2,6-diisopropylphenyl)imine (8.00 g, 33 mmol), cyclohexylamine (4.1 mL, 36 mmol), and 4-toluenesulfonic acid monohydrate (100 mg) in benzene (125 mL) was connected with a water separator and a reflux condenser. The solution was refluxed for 4 h. The solvent was evaporated under reduced pressure. Vacuum distillation gave 10.10 g (95%) of product, b.p. 118–124 °C/0.05–0.4 mm Hg. 1H NMR (CDCl3): δ = 6.88–7.16 (m, 3 H), 3.53 (m, 1 H, 3JHH = 4.7 Hz, NCH), 2.44–2.71 (m, 2 H), 2.20 (s, 3 H, CH3), 1.84 (s, 3 H, CH3), 1.90–1.23 (8 H), 1.19 (d, 6 H, 3JHH = 6.1 Hz, CH3), 1.11 (d, 6 H, 3J = 6.9 Hz, CH3). – 13C{1H} NMR (CDCl3): δ = 169.0, 164.1, 146.6, 135.1, 123.2, 122.8, 60.3, 33.4, 28.1, 25.8, 24.6, 23.1, 22.7, 16.2, 12.4.
2-Chloro-1-cyclohexyl-3-(2,6-diisopropylphenyl)-4,5-dimethyl-1,3,2-diazaphospholene 2: A piece of lithium (380 mg, 55 mmol) was added to a solution of 1-cyclohexyl-4-(2,6-diisopropylphenyl)-1,4-diazabutadiene (5.11 g, 16 mmol) in THF (15 mL). The mixture was stirred for 12 h at r.t. Remaining lithium was removed, and the deeply colored solution formed was cooled to −78 °C. Solid triethylamine hydrochloride (4.74 g, 34 mmol) was added in several portions, and the reaction mixture was allowed to warm slowly to r.t. and stirred for 4 h at this temperature. The solution was then again cooled to −78 °C, and phosphorus trichloride (1.4 mL, 16 mmol) was added dropwise via syringe. The reaction mixture was allowed to warm to r.t. and was stirred for 12 h at this temperature. The solvent was evaporated under reduced pressure, the residue dissolved in toluene (15 mL), and filtered. The filtrate was concentrated under reduced pressure until the solution became turbid, and the product was then isolated by crystallization at 4 °C; yield 3.7 g (60%, m.p. 177.0 °C). – 1H NMR (CDCl3): δ = 7.42 (t, 1 H, 3JHH = 7.8 Hz, p-CH), 7.25 (d, 2 H, 3JHH = 7.2 Hz, m-CH), 4.09–4.26 (br, 1 H, N–CH), 2.48–2.64 (m, 2 H), 2.42 (s, 3 H), 2.34–2.48 (m, 2 H), 1.86 (s, 3 H), 1.67–2.06 (m, 4 H), 1.41–1.62 (m, 2 H), 1.24–1.41 (m, 2 H), 1.20, (d, 6 H, 3JHH = 6.8 Hz, CH3), 1.13, (d, 6 H, 3JHH = 6.7 Hz, CH3). 13C{1H} NMR (CDCl3; not all quaternary carbon atoms detected due to signal broadening and insufficient signal-to-noise ratio): δ = 147.4 (i-C), 130.3 (p-C), 124.5 (m-C), 58.8 (d, 2JCP = 8 Hz, NCH), 35.2 (CH2), 35.0 (CH2), 28.3 (HC), 25.9 (CH3), 25.8 (CH2), 25.0 (CH2), 23.8 (CH3), 12.5 (CH3), 11.5 (CH3). 31P{1H} NMR (CDCl3): δ = 182.3 (s). – Calcd. for C22H34N2PCl (392.95): C 67.24 H 8.72 N 7.13; found: C 66.74 H 8.83 N 6.77.
1-Cyclohexyl-3-(2,6-diisopropylphenyl)-4,5-dimethyl-1,3,2-diazaphospholium trifluoromethanesulfonate 3: Trimethylsilyl trifluoromethanesulfonate (0.59 mL, 3 mmol) was added dropwise to a stirred solution of 2-chloro-1-cyclohexyl-3-(2,6-diisopropylphenyl)-4,5-dimethyl-1,3,2-diazaphospholene (0.86 g, 2 mmol) in toluene (5 mL). The mixture was stirred for 1 h, and all volatiles were removed in vacuum. The residue was extracted three times with diethyl ether (5 mL) and dried in vacuum (yield 1.19 g, 99%; dec. > 300 °C). 1H NMR (CDCl3): δ = 7.54 (t, 1 H, 3JHH = 7.8 Hz, p-CH), 7.34 (d, 2 H, 3JHH = 7.8 Hz, m-CH), 4.56 (quint, 1 H, 3JHH = 3.6 Hz, N–CH), 2.67 (d, 3 H, 5JPH = 1.3 Hz, CH3), 2.52–2.65 (m, 2 H, CH2), 2.29 (sept, 2 H, 3JHH = 6.8 Hz, CH), 2.08 (d, 3 H, 5JPH = 1.9 Hz, CH3), 1.89–2.04 (m, 2 H, CH2), 1.80–1.89 (m, 1 H, CH2), 1.64 (t, 4 H, 3JHH = 10.1 Hz, CH2), 1.30 (m 1 H, CH2), 1.18 (d,3JHH = 6.8 Hz, 6 H, CH3), 1.15 (d, 3JHH = 6.8 Hz, 6 H, CH3). – 13C{1H}-NMR (CDCl3): δ = 157.0 (i-C), 146.1 (o-C), 142.61 (C–N), 142.56 (C–N), 131.7 (p-C), 125.0 (m-C), 60.7 (d, 2JCP = 7.3 Hz, CH–N), 36.7 (d, 3JCP = 13.4 Hz, CH2), 28.2 (CH), 25.9 (CH3), 25.5 (CH2), 24.9 (CH2), 23.1 (CH3), 13.6 (CH3), 12.4 (CH3). – 31P{1H} NMR (CDCl3): δ = 203.8 (s). – MS (EI, 70 eV, 415K): m/e (%): 506.1 ([M]+, 20), 357.2 ([M –SO3CF3]+, 100), 275.1 ([M –SO3CF3C6H10], 17), 202.1 ([M –SO3CF3C6H11NPC2H2]+, 5). – Calcd. for C23H34N2O3PSF3 (506.56): C 54.53 H 6.77 N 5.53 P 6.11, found: C 53.51 H 7.05 N 5.33.
1-Methyl-3-alkyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonates 6a–f (general procedure): A suspension of the appropriate cyclotetra-1-alkyl-benzo-1,3,2-diazaphosphole [19] (1.1 mmol) and methyl trifluoromethanesulfonate (0.6 mL, 5.4 mmol) in xylene (15 mL) was refluxed until the solid had completely dissolved (about 1.5 h). The mixture was allowed to cool to room temperature and about three quarters of the solvent were evaporated under reduced pressure. A suspension formed from which the rest of solvent was separated from the solid phase with a syringe. The remaining solid was washed twice with hexane (5 mL) and once with hexane-diethyl ether (1:1, total volume 3 mL). The remaining product was dried in vacuum.
1,3-Dimethyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (6a): Yield 1.26 g (98%), m.p. 123.1 °C. – 1H NMR (CDCl3): δ = 7.62–7.67 (m, 4 H, CH), 4.13 (d, 3 H, 3JHP = 11.0 Hz, NCH3). – 13C{1H} NMR (CDCl3): δ = 138.6 (d, 2JCP = 6.1 Hz, C), 127.1 (d, 4JCP = 1.9 Hz, CH), 113.0 (d, 3JCP = 1.9 Hz, CH), 33.1 (d, 2JCP = 17.6 Hz, NCH3). – 31P{1H} NMR (CDCl3): δ = 215.5 (s). Calcd. for C9H10F3N2O3PS (314.22): C 34.40 H 3.21 N 8.92, found C 34.00 H 3.38 N 8.72.
1-Ethyl-3-methyl-benzo-1,3,2-diazaphospholium trifluoromethansulfonate (6b): Yield 0.80 g (86%), m.p. 82.7 °C. – 1H NMR (CDCl3): δ = 7.63–7.64 (m, 4 H, CH), 4.53 (dq, 2 H, 3JHH = 7.2 Hz, 3JHP = 11.4 Hz, NCH2), 4.12 (d, 3 H, 3JHP = 11.0 Hz, NCH3), 1.80 (dt, 3 H, 3JHH = 7.2 Hz, 4JHP = 1.0 Hz, CCH3). – 13C{1H} NMR (CDCl3): δ = 139.1 (d, 2JCP = 6.2 Hz, C), 137.8 (d, 2JCP = 6.0 Hz, C), 127.2 (d, 4JCP = 1.8 Hz, CH), 126.9 (d, 4JCP = 1.9 Hz, CH), 113.3 (d, 3JCP = 1.4 Hz, CH), 113.2 (d, 3JCP = 1.4 Hz, CH), 42.9 (d, 2JCP = 14.6 Hz, NCH2), 33.3 (d, 2JCP = 17.7 Hz, NCH3), 15.5 (d, 3JCP = 10.9 Hz, CCH3). – 31P{1H} NMR (CDCl3): δ = 215.9 (s). – Calcd. for C10H12F3N2O3PS (328.25): C 36.59 H 3.68 N 8.53, found C 37.14 H 3.63 N 8.62.
1-Methyl-3-propyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (6c): Yield 1.34 g (87%), m.p. 95.5 °C. – 1H NMR (CDCl3): δ = 7.61–7.65 (m, 4 H, CH), 4.43 (m, 2 H, NCH2), 4.14 (d, 3 H, 3JHP = 11.0 Hz, NCH3), 2.16 (m, 2 H, CCH2), 1.12 (t, 3 H, 3JHH = 7.4 Hz, CH3). – 13C{1H} NMR (CDCl3): δ = 139.0 (d, 2JCP = 6.3 Hz, C), 137.7 (d,, 2JCP = 5.5 Hz, C), 127.2 (d, 4JCP = 1.8 Hz, CH), 126.9 (d, 4JCP = 1.8 Hz, CH), 113.3 (d, 3JCP = 1.4 Hz, CH), 113.2 (d, 3JCP = 1.4 Hz, CH), 49.1 (d, 2JCP = 13.3 Hz, NCH2), 33.3 (d, 2JCP = 17.5 Hz, NCH3), 23.4 (d, 3JCP = 8.6 Hz, CCH2), 11.3 (d, 4JCP = 1.7 Hz, CCH3). – 31P{1H} NMR (CDCl3): δ = 215.8 (s). – Calcd. for C11H14F3N2O3PS (342.27): C 38.60 H 4.12 N 8.18, found C 39.09 H 3.98 N 8.13.
1-Butyl-3-methyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (6d): Yield 0.72 g (92%), m.p. 80.8 °C. – 1H NMR (CDCl3): δ = 7.61–7.65 (m, 4 H, CH), 4.46 (dt, 2 H 3JHH = 7.5 Hz, 3JPH = 10.7 Hz, NCH2), 4.14 (d, 3 H 3JHP = 11.0 Hz, NCH3), 2.10 (quint, 2 H 3JHH = 7.5 Hz, NCCH2), 1.53 (sext, 2 H, 3JHH = 7.4 Hz, NCCCH2), 1.01 (t, 3 H, 3JHH = 7.3 Hz, CH3) 13C{1H} NMR (CDCl3): δ = 139.1 (d, 2JCP = 6.9 Hz, C), 137.8 (d, 2JCP = 5.5 Hz, C), 127.2 (d, 4JCP = 1.8 Hz, CH), 126.9 (d, 4JCP = 1.8 Hz, CH), 113.3 (d, 3JCP = 1.3 Hz, CH), 113.2 (d, 3JCP = 1.5 Hz, CH), 47.3 (d, 2JCP = 13.3 Hz, NCH2), 33.3 (d, 2JCP = 17.4 Hz, NCH3), 31.9 (d, 3JCP = 8.4 Hz, NCCH2), 20.1 (d, 4JCP = 1.7 Hz, NCCCH2), 13.4 (s, CCH3). – 31P{1H} NMR (CDCl3): δ = 216.0 (s). – Calcd. for C12H16F3N2O3PS (356.30): C 40.45 H 4.53 N 7.86, found C 40.11 H 4.62 N 7.76.
1-Methyl-3-pentyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (6e): Yield 0.93 g (67%), m.p. 97.5 °C. – 1H NMR (CDCl3): δ = 7.61–7.65 (m, 4 H, CH), 4.45 (dt, 2 H, 3JHH = 7.6 Hz, 3JPH = 10.7 Hz, NCH2), 4.13 (d, 3 H, 3JHP = 11.0 Hz, NCH3), 2.11 (quint, 2 H, 3JHH = 7.4 Hz, NCCH2), 1.39–1.47 (m, 4 H, CH2), 0.92 (t, 3 H, 3JHH = 7.0 Hz, CH3). – 13C{1H} NMR (CDCl3): δ = 139.1 (d, 2JCP = 6.3 Hz, C), 137.8 (d, 2JCP = 6.1 Hz, C), 127.2 (d, 4JCP = 1.8 Hz, CH), 126.9 (d, 4JCP = 1.8 Hz, CH), 113.4 (d, 3JCP = 1.3 Hz, CH), 113.2 (d, 3JCP = 1.5 Hz, CH), 47.6 (d, 2JCP = 13.3 Hz, NCH2), 33.3 (d, 2JCP = 17.5 Hz, NCH3), 29.7 (d, 3JCP = 8.5 Hz, NCCH2), 28.9 (d, 4JCP = 1.6 Hz, NCCCH2), 22.0 (s, CH2), 13.7 (s, CH3). – 19F NMR (CDCl3): δ = −78.5 (s). – 31P{1H} NMR (CDCl3): δ = 216.0 (s). – Calcd. for C13H18F3N2O3PS (370.33): C 42.16 H 4.90 N 7.56, found C 41.65 H 4.97 N 7.36.
1-Hexyl-3-methyl-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (6f): Yield 0.75 g (60%), m.p. 100.5 °C. – 1H NMR (CDCl3): δ = 7.61–7.65 (m, 4 H, CH), 4.45 (dt, 2 H, 3JHH = 7.6 Hz, 3JHP = 10.7 Hz, NCH2), 4.14 (d, 3 H, 3JHP = 11.0 Hz, NCH3), 2.11 (quint, 2 H, 3JHH = 7.5 Hz, NCCH2), 1.28–1.53 (m, 6 H, CH2), 0.89 (t, 3 H, 3JHH = 7.1 Hz, CH3). – 13C{1H} NMR (CDCl3): δ = 140.0 (d, 2JCP = 5.9 Hz, C), 137.1 (d, 2JCP = 5.0 Hz, C), 127.2 (d, 4JCP = 1.8 Hz, CH), 126.9 (d, 4JCP = 1.9 Hz, CH), 113.3 (d, 3JCP = 1.3 Hz, CH), 113.2 (d, 3JCP = 1.4 Hz, CH), 47.6 (d, 2JCP = 13.2 Hz, NCH2), 33.3 (d, 2JCP = 17.4 Hz, NCH3), 31.0 (s, CH2), 29.9 (d, 3JCP = 8.6 Hz, NCCH2), 26.6 (d, 4JCP = 1.6 Hz, NCCCH2), 22.3 (s, CH2), 13.8 (s, CH3). – 19F NMR (CDCl3): δ = −78.5 (s). – 31P{1H} NMR (CDCl3): δ = 215.9 (s). – Calcd. for C14H20F3N2O3PS (384.35): C 43.75 H 5.24 N 7.29, found C 43.03 H 5.31 N 6.87.
1,3-benzo-1,3,2-diazaphospholium trifluoromethanesulfonate (8) [21]: The synthesis was carried out by analogy to that of the 1,3-dialkyl derivatives except that a solution of the appropriate amount of trifluoromethanesulfonic acid in 10 mL xylene was used instead of the methyl ester and that the solid remaining after evaporation of the solvent was washed three times with diethyl ether. Cooling of the washing liquid to −20 °C produced a further quantity of product in the form of colorless crystals (m.p. 132.0 °C) which were suitable for single-crystal X-ray diffraction studies. The combined yield was 0.84 g (99%). 1H NMR (CD3CN): δ = 12.50 (broad, 2 H, NH), 7.53–7.90 (m, 4 H, CH). – 13C{1H} NMR (CD3CN): δ = 137.5 (d, 2JCP = 6.7 Hz, C), 127.3 (d, 4JCP = 1.9 Hz, CH), 115.3 (d, 3JCP = 1.2 Hz, CH)́. – 31P{1H} NMR (CD3CN): δ = 212.0 (s). – Calcd. for C7H6F3N2O3PS (286.17): C 64.84 H 8.16 N 12.60; found C 64.84 H 8.35 N 12.61.
Crystallography. The crystal structure determinations of 2 and 6d,f were performed on Bruker-Nonius Kappa-CCD diffractometers at 100(2) K (2, 6d) or 123(2) K (6f), respectively, using Mo-Kα radiation (λ = 0.71073 Å). Direct Methods (SHELXS-97) [25] were used for structure solution, and refinement was carried out using SHELXL-97 (full-matrix least-squares on F2) [25]. Hydrogen atoms were refined using a riding model. The studied crystal of 3 was a non-merohedral twin with two domains (80:20; BASF 0.206(2)), twin-matrix: (−1 0 0, −0.108 1 −0.020, 0 0 −1) [26].
2: colorless crystals, C22H34ClN2P, M = 392.93, crystal size 0.20 × 0.10 × 0.03 mm, triclinic, space group , a = 11.1169(3) Å, b = 13.2635(4) Å, c = 14.7574(4) Å, α = 89.127(2)°, β = 84.693(2)°, γ = 87.350(2)°, V = 2164.19(11) Å3, Z = 4, ρ(calcd) = 1.206 Mg/m3, F(000) = 848, μ = 0.26 mm−1, 7571 reflections (2θmax = 50°), 7571 unique [Rint = 0.000], 474 parameters, GooF = 1.16, R1 (I > 2σ(I)) = 0.079, (all data) = 0.212, largest diff. peak and hole 0.496/−0.463 e A−3.
6d: colorless crystals, C12H16F3N2O3PS, M = 356.30, crystal size 0.40 × 0.40 × 0.20 mm, triclinic, space group P̄1 (No. 2), a = 6.842(4) Å, b = 8.792(5) Å, c = 13.740(7) Å, α = 78.49(4)°, β = 85.24(2)°, γ = 84.24(2)°, V = 804.2(7) Å3, Z = 2, ρ(calcd) = 1.471 Mg/m3, F(000) = 368, μ = 0.34 mm−1, 3438 reflections (2θmax = 52°), 3160 unique [Rint = 0.027], 201 parameters, no restraints, GooF = 0.961, R1 (I > 2σ(I)) = 0.040, (all data) = 0.1086, largest diff. peak and hole 0.377/−0.330 e A−3.
6f: colorless crystals, C14H20F3N2O3PS, M = 384.35, crystal size 0.40 × 0.10 × 0.05 mm, monoclinic, space group P 21/c (No. 14), a = 15.017(2) Å, b = 8.913(1) Å, c = 13.245(1) Å, β = 98.21(1)°, V = 1754.6(3) Å3, Z = 4, ρ(calcd) = 1.455 Mg/m3, F(000) = 800, μ = 0.32 mm−1, 14489 reflections (2θmax = 50°), 3086 unique [Rint = 0.094], 217 parameters, GooF = 1.03, R1 (I > 2σ(I)) = 0.057, (all data) = 0.143, largest diff. peak and hole 0.401/−0.288 e A−3.
CCDC-762960 (2), CCDC-763485 (6d), and CCDC-762959 (6f), contain the supplementary (Supplementary data) crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
We thank J. Trinkner and K. Wohlbold (Institut für Organische Chemie, University of Stuttgart) for recording the mass spectra and C. Schneck (Institut für Anorganische Chemie, University of Stuttgart) for thermoanalytical measurements. The Deutsche Forschungsgemeinschaft is acknowledged for financial support. D.F. thanks further the DAAD, D.G. COST (action CM0802), and M.N. the Academy of Finland (proj. no. 128800) for financial support.
1 The limitation is in fact introduced by the restricted availability of isolable ketimines needed as precursors to 1; in attempting to prepare such derivatives from sterically less demanding N-aryl amines or more basic N-alkyl amines, we obtained only inseparable mixtures of the desired ketimines with the corresponding symmetrical α-diimines that were useless for the further synthesis.
2 A further example is exemplified in the isolation of a parent benzazathiaphospholium salt [22].