

Phosphorus derivatives with six substituents at the immediate periphery of the P-atom are quite common, more than usually imagined [1–7]. Many derivatives of varied charge and geometry have been synthesized in studies devoted to phosphorus reactivity. A large number of compounds have also been prepared for applications in fields as varied as classical organic chemistry, stereoselective synthesis and analysis [8,9], bioorganic chemistry, electrochemistry and photochemistry [10]. This personal account will, however, concentrate its attention on a single aspect of this chemistry, that of the anionic hexacoordinated phosphates – and on their use as chiral NMR solvating, resolving and asymmetry-inducing reagents, in particular.
In fact, many chemical reactions and processes involve cationic reagents, intermediates or products. Cations are frequent intermediates along reaction pathways that react with nucleophiles to produce interesting fragments and functional groups. Cations are often Lewis acidic and numerous applications have been developed using such reagents. A large range of important synthetic and biological processes are mediated by nitrogen containing cations. Cations are also efficient templates for the construction of complex supramolecular arrays, such as the catenanes, knots, helicates, rotaxanes, molecular muscles and presses. Cations have also interesting physical properties being often synthetic dyes with many applications.
For the purpose of this article, cations can also be prochiral or chiral and many of their applications, reactions or processes lead to racemic molecular or supramolecular assemblies. To afford instead non-racemic or enantiopure products, and benefit from possible new applications, an asymmetric ion pairing with chiral unreactive anionic counterions can be considered – the gegenions behaving as remote asymmetric auxiliaries1 [11]. This field of chiral anion mediated ion pairing chemistry was reviewed previously – the latest extensive reports dating from 2003 and 2009 [12,13]. Only selected examples will therefore be reported in this manuscript along with, sometimes, a more personal view on some of the aspects.
1 Chiral hexacoordinated phosphate anions
The octahedral geometry of pentavalent hexacoordinated phosphorus allows indeed the formation of chiral anions by complexation of a central phosphorus atom with three identical dianionic bidentate ligands. As early as 1965, Hellwinkel reported the synthesis of hexacoordinated phosphate anion 1 which was shown to be chiral and configurationally stable through a resolution procedure [14–16]. The enantiomers exist as Λ or Δ antipodes with left- and right-handed propeller shape (M or P helicity) respectively [17,18]. Hellwinkel also prepared in the 1970s several other anionic phosphates and that combining around the octahedral P atom three benzenediolato ligands (e.g., 2) [19,20], or combinations of catecholato and bidentate biphenylidene chelating moieties (3 and 4 in particular, Fig. 1) [19]. These anions were, however, studied only in racemic series for reasons that will become obvious in the following paragraphs.
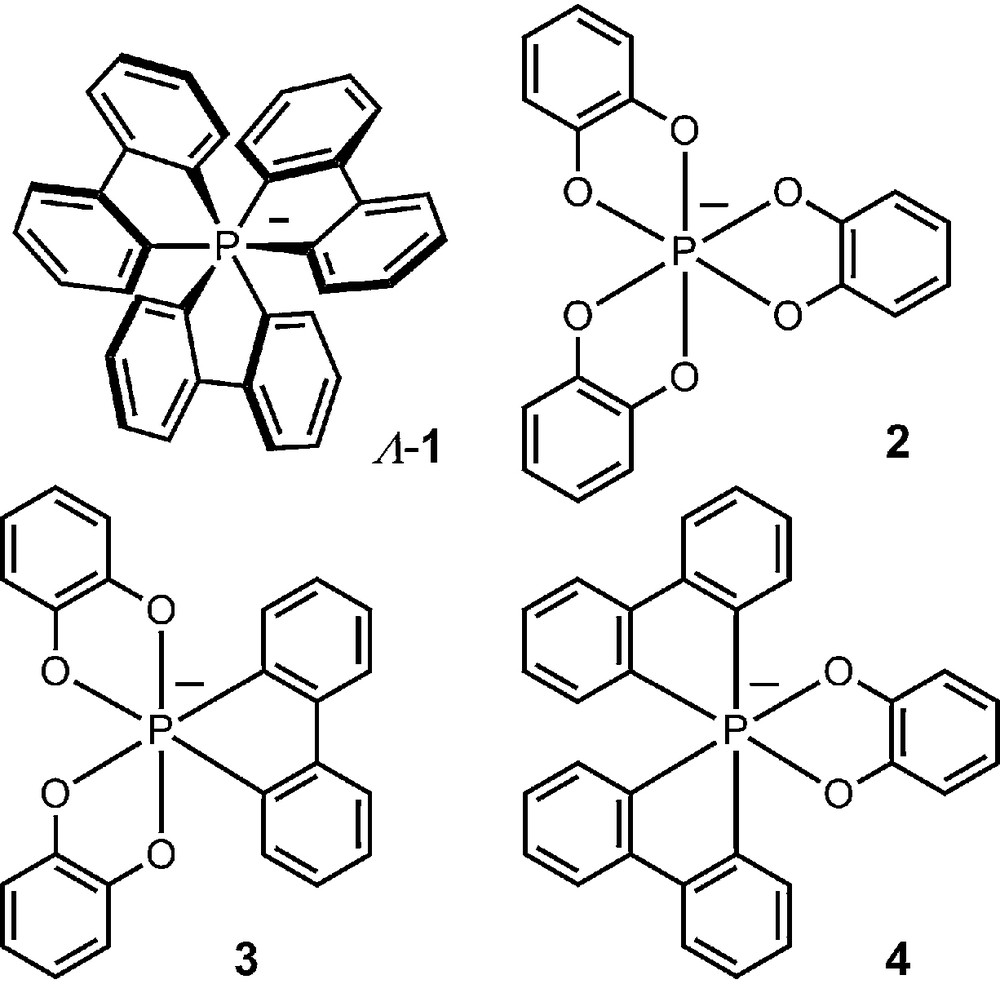
Example of early hexacoordinated phosphates.
Indeed, a decade later, Wolf, Koenig et al. studied in more details tris(benzenediolato)phosphate anion 2, of particular interest for its easy one-step preparation from catechol, PCl5 and an amine. It was then shown that anion 2 racemizes unfortunately rapidly in solution as an ammonium salt [21–24]. Mechanistic studies demonstrated further that the racemization of 1 was acid-catalyzed and followed first order in substrate rate reaction law. An irregular intramolecular one-ended dissociation mechanism was proposed to explain the results (Scheme 1): protonation-induced ring opening of 2 to a bipyramidal spirophosphorane intermediate 5, followed by pseudorotation of the substituents around the phosphorus and random recyclisation result in the observed loss of chirality. Determination of the kinetic parameters (ΔH≠ = 20.3 kcal mol−1, ΔS≠ = –4.1 kcal mol−1 K−1) led the authors to suggest the ring opening to the phosphorane as the rate-determining step. This observation of a poor configurational stability for 2 in solution resulted in an overall lack of interest for this type of compounds from the stereochemical community – and this for more than two decades.
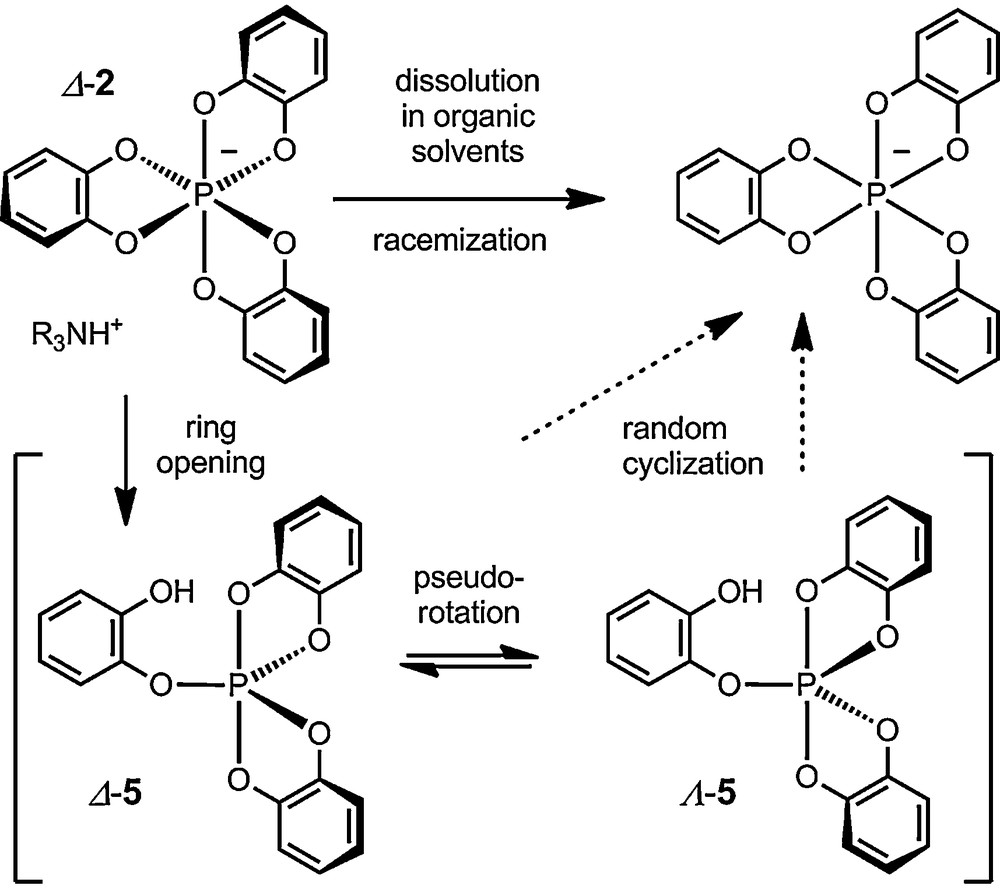
Racemization of ammonium tris(benzenediolato)phosphates in solution. Anion 2 is arbitrary represented with a Δ absolute configuration.
However, this chemistry of hexacoordinated phosphate anions was recently rejuvenated as chiral tris(tetrachlorobenzenediolato)phosphate(v) anion 6 was shown to be configurationally stable at room temperature in all common organic solvents [25]. The realization that this anion would be configurationally stable came from two different perspectives.
One is theoretical and is based on the mechanism detailed above in Scheme 1. We realized in 1996 that any decrease of the basicity of the oxygen atoms of a phosphate moiety of type 2 should reduce or avoid their protonation, consequently stop the opening to the phosphorane and hence avert the racemization. Configurationally stable hexacoordinated phosphate anions should result from the use of catechols substituted with electron-withdrawing groups – such as tetrachlorocatechol2.
The second perspective is a practical one and comes from the chemistry from Schmutzler et al. [26]. In 1992, the Braunschweig group reported the oxidation of diazadiphosphorinanone 7 with two equivalents of o-chloranil 8. This reaction leads to the cleavage of the original heterocycle and to the formation of a mixture of compounds, including 1,3,2-diazaphosphetidine 9 and spirophosphorane 10. The key for the development of our own chemistry was the mention in this report that compound 9 undergoes a slow but spontaneous and quantitative transformation in chloroform to anion 6. It was an excellent indication of the thermodynamic stability of the anion and, for us, of a possible configurational permanence as well.
This D3-symmetric anion 6, known as TRISPHAT (Schemes 2 and 3) is usually prepared using a procedure similar to those historically developed by Hellwinkel and Koenig – that is, the one-time addition of three equivalents of tetrachlorocatechol to one equivalent of PCl5 to afford the hexacoordinated phosphate anion [25]. The formation of the P(VI) anion is easily driven to completion by the addition of a base to the medium such as an aliphatic amine – usually nBu3N. The racemic anion can then be resolved by association with a N-H-cinchonidinium as cationic counterion. Both Δ and Λ enantiomers can be isolated on large scale as [cinchonidinium][Δ-6] and [Bu3NH][Λ-6] salts respectively [27].
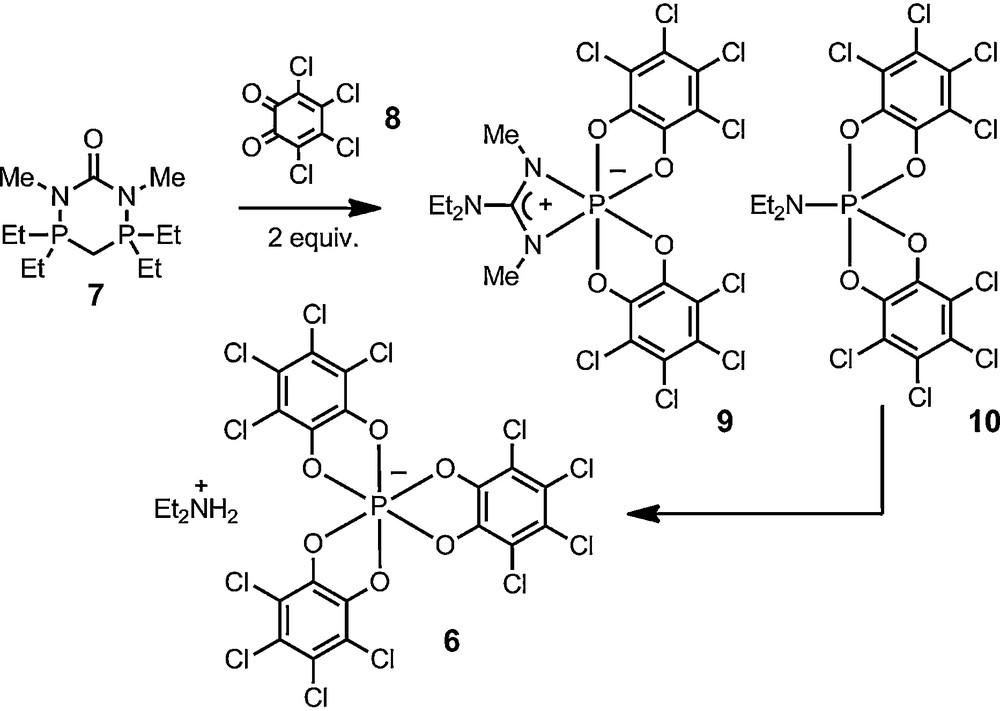
Schmutzler's report of the spontaneous formation of anion 6.
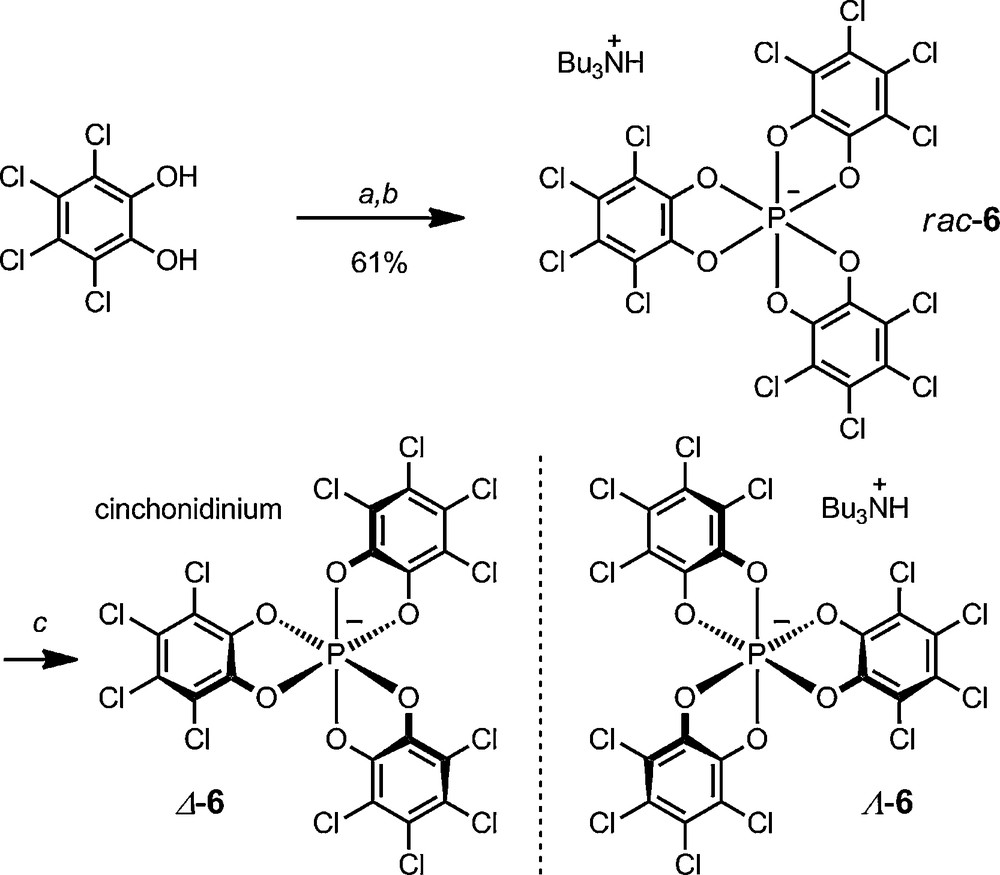
Synthesis and resolution of TRISPHAT anion 6. (a) PCl5 (0.33 equiv.), Ph-Me, 70 °C. (b) nBu3N, CH2Cl2 / n-Hexane, 20 °C. (c) cinchonidine (0.50 equiv.), CH2Cl2, 20 °C.
The synthesis of functionalized/mixed hexacoordinated phosphate anions containing two tetrachlorocatechols and a third different 1,2-diolato ligand can also be achieved. However, the preparation of such P(VI) anions is not a trivial matter [28]. A practical and general synthetic procedure had to be developed for the making of such novel hexacoordinated phosphates; anion BINPHAT 11 (Scheme 4), which includes a BINOL moiety, is the archetype of such derivatives [29]. The procedure for the making of these advanced anions involves the in situ (and voluntary) formation of the spirophosphorane intermediate 10 (Scheme 2), which reacts with essentially any diols to generate, in one-pot, mixed hexacoordinated phosphate anions. For instance, BINPHAT anion 11 can be prepared in good yields (70–85% yield) from tetrachlorocatechol, HMPT, o-chloranil 8 and BINOL. This protocol was recently streamlined – all reagents being added as essentially one equivalent. It can be scaled-up to multi-gram quantities [30]. Starting from enantiopure R- or S-BINOL, the diastereoselectivity of the reaction is high (> 96%) and yields are reproducible in favor of Λ-11 or Δ-11 respectively; the stereoselectivity being most probably of kinetic rather than thermodynamic origin [31].
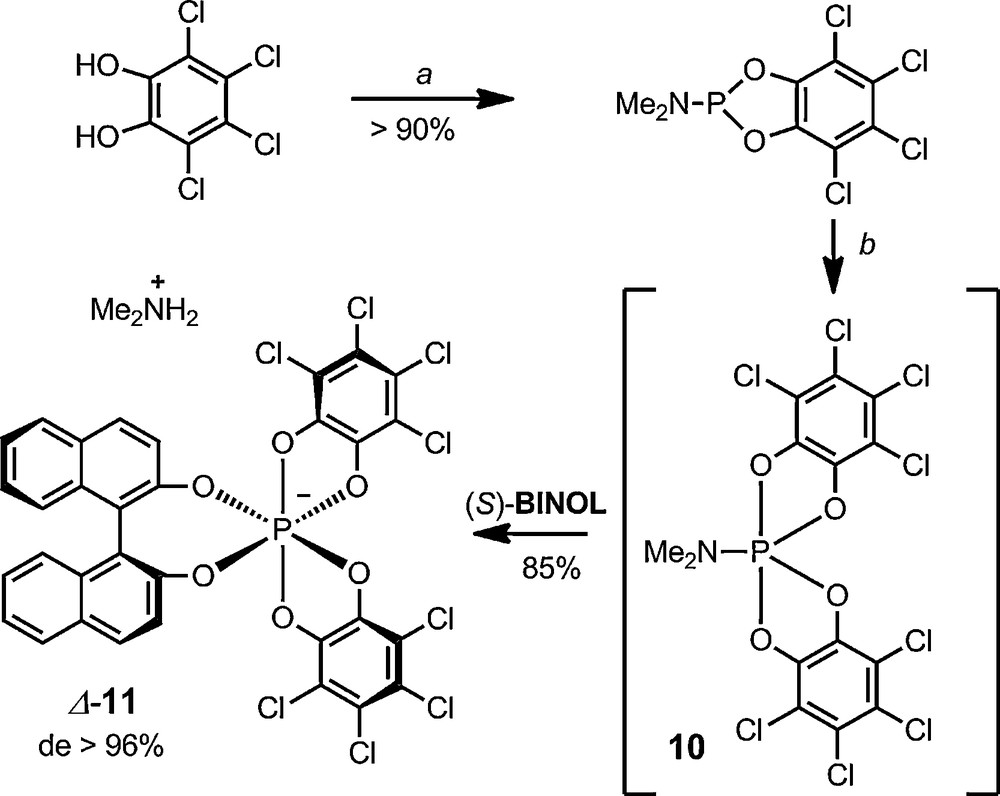
One-pot synthesis of BINPHAT anion 11. (a) HMPT, toluene, reflux. (b) o-chloranil (1.0 equiv.), BINOL (1.0 equiv.), CH2Cl2, 20 °C.
It is feasible, although with some risks, to change the oxidant (o-chloranil) in this process for another ortho-quinone, but original phosphate anions such as phenanthroline derivative 12 can result from the operation (see below) [28]. The one-pot procedure can be extended to the making of other mixed phosphates (13–16) by varying the nature of the diol added last in the protocol (e.g., sugar diols, tartrate, dihydrobenzoin derivatives, fluorocatechols) [28,30,32–34].
Anions 16a and 16b contain fluorine substituents at selected positions. They display unique NMR properties that can be used for the determination of exact (absolute) ion pairing structures in solution. For the resolution of these two anions, a novel and general protocol was developed using N-benzyl-cinchonidium chloride as a resolving agent. The Δ and Λ enantiomers of the anions are afforded in very high chemical and enantiomeric purity [33] (Fig. 2).
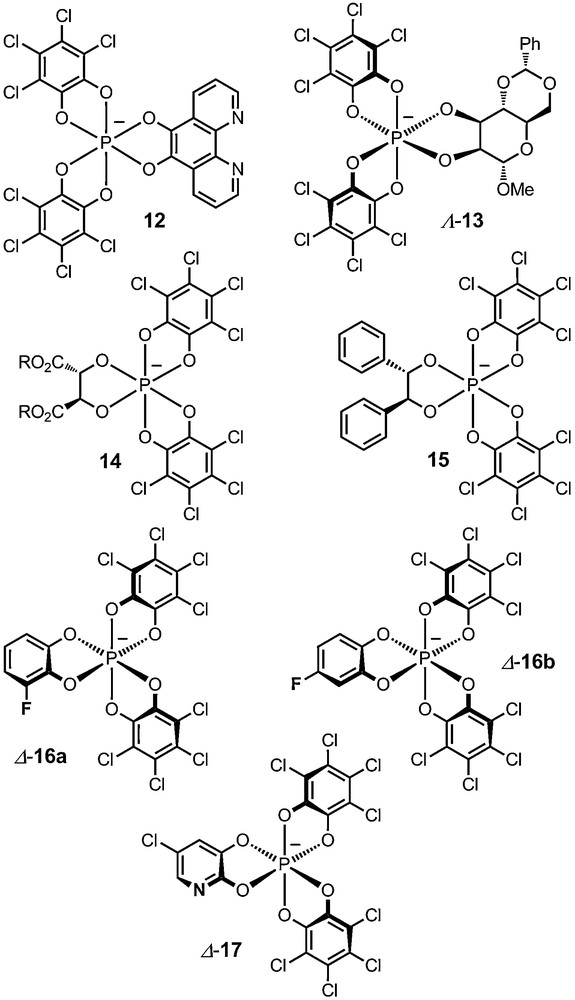
Functionalized hexacoordinated phosphates 12–17.
Finally, the synthesis of a novel nitrogen-containing hexacoordinated phosphate anion 17, named TRISPHAT–N, was very recently reported. This unique anion, through its Lewis basic nitrogen atom, can interact directly with metal centers and allow the stereocontrol of molecular events that previous non-coordinating anions of the TRISPHAT family could not achieve. It can, for instance, control the absolute P or M configuration of axially chiral tropos ligands bound to metal centers or the configuration of metal centers themselves (d.r. up to 96:4) [35]. Other applications in catalysis have been developed and will be mentioned later.
2 Successful NMR chiral solvating agents
As already mentioned, chiral cations are involved in many areas of chemistry and, unfortunately, only a few simple methods are available to determine with precision their enantiomeric purity. In the last decades, NMR has evolved as one of the methods of choice for the measurement of the enantiomeric purity of chiral species [36–41]. Neutral chiral lanthanide shift reagents, which are particularly efficient for most applications, are however rarely used with chiral cations due to an overall lack of interactions with these analytes that often miss accessible electron donor sites. Anionic substances have therefore an advantage over neutral reagents to behave as NMR chiral solvating agents for chiral cations. They can form contact pairs and the short-range diastereomeric interactions that result can lead to clear differences in the NMR spectra of the two diastereoisomeric salts. Several types of applications have made use of this effect.
2.1 Evidencing molecular chirality (and prochirality)
The first type of application is the use of TRISPHAT 6 and BINPHAT 11 anions as general NMR chiral solvating anions to evidence molecular chirality. The cationic moieties of study can be of organic, organometallic or metalloorganic nature. The stereogenic elements in the analytes can be of central, axial, planar or helical chirality. 1H, 13C, 15N, 19F and 31P NMR spectroscopy have been used in these studies. In Fig. 3 are represented some of the cationic derivatives that have been analyzed with success with these reagents over the last ten years or so. Other systems are reported in the next sections or in the following references [42–45].
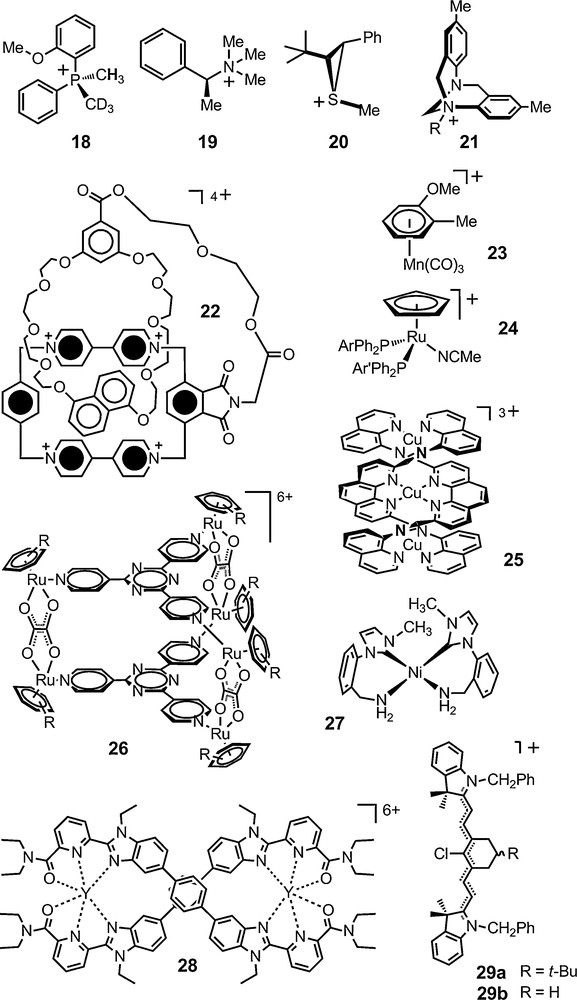
Chiral cationic assemblies analyzed successfully with anions 6 and 11.
BINPHAT 11 has often-superior chiral solvating properties with organic cations (e.g., 18–20) [46–49]. Interestingly, the enantiomers of N-alkyl Tröger salts of type 21 are readily distinguished – even in polar solvents such as CD3CN [50]. Planar chiral donor-acceptor pretzelanes of type 22 can also be analyzed with this anion as shown by Stoddart recently [51]. TRISPHAT 6 is overall more efficient with cationic metallo-organic and organometallic substrates [52–55]. It is the case for manganese planar-chiral and ruthenium chiral-at-metal arene complexes 23 and 24, tricopper(I) helicate 25, a chiral tetrairon(II) square complex, cationic triangular metallo-prisms of type [Ru6(arene)6(tpt)2(C2O4)3]6+ 26 (arene = C6Me6 and p-PriC6H4Me; tpt = 2,4,6-tripyridyl-1,3,5-triazine), bisporphyrin octanuclear metalla-boxes, and chiral pseudotetrahedral cationic cages as well [56–63].
Very recently, a homoleptic nickel(II) complex 27 with primary amino-functionalized N-heterocyclic carbene ligands was prepared by the reduction of a nitrile-functionalized imidazolium salt. This square-planar nickel(II) complex was characterized by NMR spectroscopy in the presence of anion 6 that helped evidenced the axial chirality of the cation. Lanthanide-containing double-stranded helicate 28 was also analyzed in the presence of this anion, its enantiodifferentiation ability being used by the authors to confirm in solution the existence of two enantiomeric forms of D2-symmetry [64,65].
Finally, anion 6 was used in conjunction with prochiral heptamethine cyanine dyes by Maury et al. The ion pairs were studied in non-dissociating solvents. Interestingly, two sets of signals were observed for the olefinic protons of the tert-butyl derivative 29a whereas such a difference was not seen for the simple cyclohexyl derivative 29b. Based on a X-ray study, the authors propose that the anion interacts non symmetrically with the cation and is able to distinguish the now diastereotopic faces of dye 29a – and hence prochirality rather than chirality per se [55].
2.2 Enantiomeric purity determination
Once molecular chirality evidenced on a racemic substrate, the method can of course be applied to non-racemic materials and for enantiomeric purity determination in particular. This has been the case with a series of organic and organometallic substrates. Jodry and Mikami have used anion 6 for assuring the enantiomeric purity of racemization-prone imidazolium cation 30 [66]. Gruselle has used the same anion to analyse the series of optically active ferrocenic salts 31 (R = Me, Et, nPr, nBu) having a planar chirality. Baseline-to-baseline separation could be readily achieved for the singlet signal of the non-substituted cyclopentadienyl ring, allowing the authors to establish a 95% enantiomeric purity for the salts [67]. Recently, this use was extended to metalloorganic complexes made of chiral enantiopure ligands. Constable et al. used salt [Et4N][Δ-6] to confirm the diastereomeric purity of an Fe(II) complex made of a C2-symmetric hexadentate ligand based on a 1,1-binaphthalene scaffold [68] (Fig. 4).
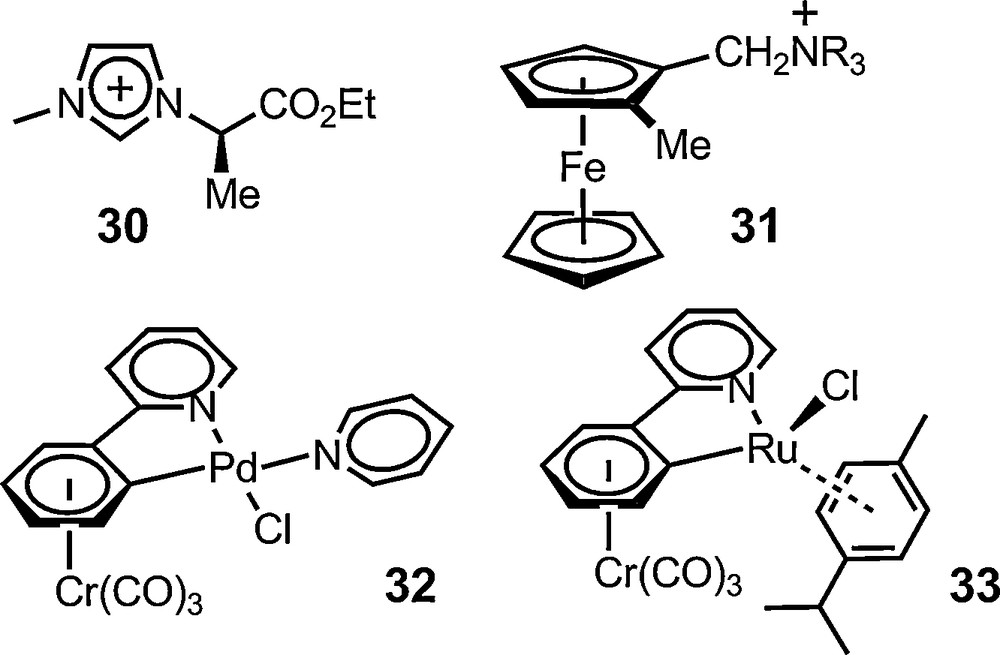
Non-racemic chiral entities analyzed with 6 and 11.
Anion 6 was also used recently by Djukic and Pfeffer with planar chiral cyclopalladated (η6-arene)Cr(CO)3 complex 32. An effective enantiodifferentiation was achieved in 20% d6-acetone and d6-benzene suggesting that the enantiomers of 32 were obtained in enantiomeric purity higher than 96% [69]. Interestingly, anion 6 failed to perform with related ruthenium complex 33. In this case, a use of salt [Bu4N][Δ-11] was necessary and it was shown that the (+)- and (–)-33 enantiomers were accessible in 82% and 89% (±2%) enantiomeric purity respectively [70]. These results broaden the fields of application of the anionic reagents to neutral species.
2.3 Determination of configurational stability
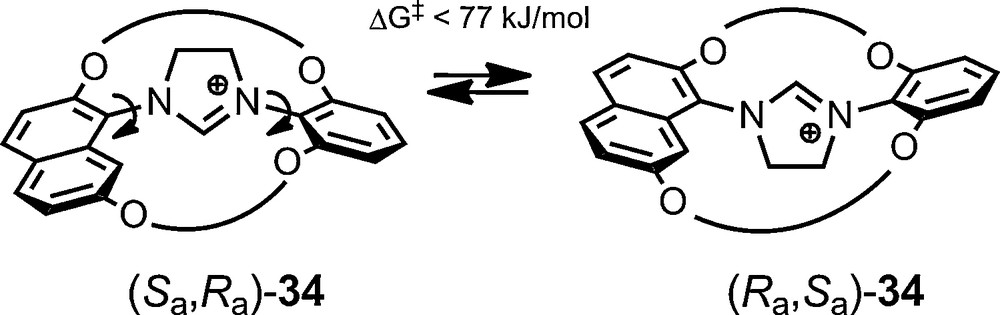
The use of anionic NMR chiral solvating reagents is, however, not limited to configurationally stable entities. Recently, in the context of a study on chiral concave N-heterocyclic carbenes (NHCs), it was shown by Lühning et al. that their bimacrocyclic imidazolinium precursors of type 34 are axially chiral. These moieties, with one symmetrically substituted benzene bridgehead and one naphthalene bridgehead, are unfortunately configurationally labile; stereodynamics being evidenced by NMR spectroscopy using 11 as stereochemical probe [71].
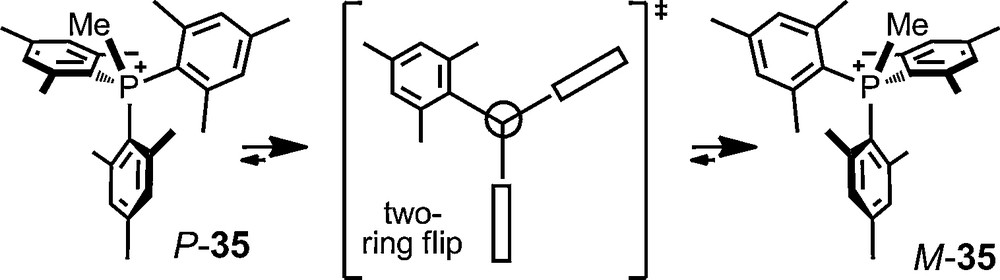
Recently, the P ⇆ M propeller isomerism of trimesitylmethylphosphonium cation 35 was also investigated using phosphate anions 6 and 11. Salts 35 [Δ-6] and 35 [Δ–11] were prepared and an enantiodifferentiation was evidenced both in 1H and 31P NMR spectroscopy (Δδmax ∼0.60 ppm, P+CH3, toluene-d8) at room temperature. The dynamic conformational isomerism among the enantiomers was detected at elevated temperatures by a broadening of the signals and characterized by a series of spectra at different temperatures (ΔG‡ ∼ 17.7 kcal mol−1) [72].
2.4 Determination of molecular symmetry
Perhaps more importantly, this asymmetric ion pairing of cation 35 with anions 6/11 allowed the first experimental evidence for the transition state structure in solution of the enantiomerization process. This was achieved by the simultaneous determination of the rates of diastereotopic group exchange and of enantiomerization; their comparison allowing the authors to conclude on a “two-ring flip” structure [72].
Recently, cationic Pd(II) allyl complexes of type 36 containing the configurationally labile ligand 1,2-bis-[4,5-dihydro-3H-dibenzo[c-e]azepino]ethane were studied as models for intermediates in Pd-catalyzed allylic alkylations. A distinction between CS- (meso) and C2-conformations was feasible using the enantiodifferentiation provided by anion 11. A preference for the CS structure was evidenced as all enantiotopic nuclei became diastereotopic in presence of 11 whereas the achirotopic atoms residing in the mirror plane did not split3 [73].
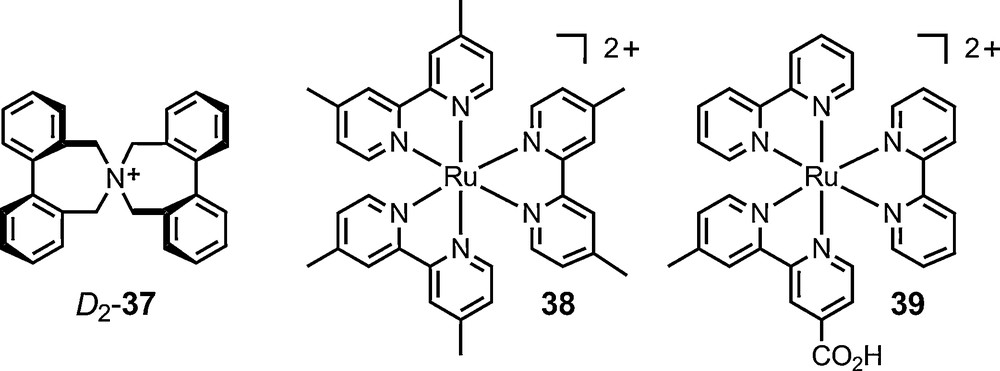
A similar approach had been used previously of the analysis of the preferred conformation (D2 vs. S4) of cation 37 (Fig. 7) using this time 15N-NMR spectroscopy [74].
Cations with precised ion pairing situations.
2.5 Precise description of ion pairing situation
Detailed structural analyses of ion pairs have shed new light on the precise nature of salt effects [75]. Much progress has been made using NMR spectroscopy. On one hand, 1H,1H-NOESY and 1H,19F-HOESY experiments have allowed qualitative and quantitative structural investigations on the interactions of cationic moieties and their counterions. On the other hand, diffusion data from pulse-field gradient spin echo (PGSE) experiments have given useful information on the average dimensions of salt aggregates in solution [76,77]. These two sets of techniques were applied to salts of chiral anionic counterions – and 6, 11 and 16 in particular. In one instance, diastereomeric salts of cation 37 [74] and Δ-11 were analyzed by 1H, 19F and 31P NMR spectroscopy and the first example of the dependence of a diffusion value on a diastereomeric relationship was demonstrated by Pregosin et al. [78] (Fig. 7).
In another instance, anions 16a and 16b were shown to possess, thanks to the precise positioning of H and F-atoms at their periphery, the ability to determine exact (absolute) three-dimensional ion pairing structures in solution. In the case of D3-symmetric dication 38, it was shown by HOESY (and NOESY) that these anions interact strongly and precisely along the C3-axis of 38 when an homochiral association is achieved [33]. Finally, Correia et al. have shown that a similar conclusion can be reached using ions 6 and 39 as analytes. Detailed PGSE and Job's titrations were performed to determine a preferred 1/2 stoichiometry of interaction and equilibrium constants were calculated for the homochiral and heterochiral salts; all data pointing towards a preferred homochiral sandwich of dication 39 sliced between two anions 6 [79].
3 Efficient resolving agents
Resolution – that is the physical separation of the enantiomers of a chiral racemic substance – has also been strongly studied with chiral anionic counterions [80,81] and only recent examples of such processes will be presented. The substrates have been racemic mixtures of chiral cations and the enantiopure anions were used as resolving agents. Upon association, attempts to separate the resulting diastereomeric ion pairs were made and, on the whole, the vast majority of the resolutions have been based on solubility differences of solids. Chromatography or asymmetric extractions were also viable possibilities.
3.1 By solubility differences of solids
Dicationic ruthenium(II) complexes of type [Ru(Me2Phen)2(NCMe)2]2+ 40a, which are important chiral building blocks in the fields of stereospecific metal complex synthesis and catalysis, can be for instance resolved with TRISPHAT 6 anions. Selective precipitation of the heterochiral salts occur, e.g. [Λ-40a][Δ-6]2, allowing a recovery of the antipodal enantiomer of the cationic complex in the mother liquor as a PF6 salt, e.g. [Δ-40a][PF6]2 [82]. Interestingly, if the resolution is attempted under irradiation, the stereoselective precipitation is then coupled to a light-induced enantiomerization of the complex leading to quantitative isolation of a single diastereomeric species [83].
This anion was also used for the resolution of organometallic complexes of rather different nature. Amouri resolved planar chiral dinuclear ruthenium complex 41 and supramolecular triangular metallo-macrocycle 42 [84–86], while Gebbink separated planar chiral ECE’-ruthenium-palladium metal complexes 43a and 43b [87] by fractional crystallization in the presence of an enantiopure TRISPHAT salt. Finally, the enantiomers of cationic [4]-helicene 44 were separated by selective precipitation using BINPHAT anion 11 [88].
3.2 By chromatography
Preparative chromatographic resolution procedures have overall freed chemists from the constraint of dependency on crystallization. They are most often performed with covalent diastereomer mixtures but ionic salts can also be separated. Whereas, historically, most ion-pair chromatographic resolutions have involved the use of enantiopure counterions in the mobile phase, recent examples have used preformed ionic diastereomer mixtures for chromatographic separations [54,89,90]. Recently, fractions of diastereomeric pairs [Λ-40b][Δ-6]2 and [Δ-40b][Δ-6]2 were readily isolated by chromatography on neutral alumina (eluent CH2Cl2); the separation requiring only a stoichiometric amount of TRISPHAT salt added to the racemic [Ru(Phen)2(NCMe)2][TfO]2 40b in acetonitrile [91]. In another study, this anion was shown to be an effective resolving agent for cyclometallated iridium dinuclear complex [(ppy)2Ir(μ-L)Ir(ppy)2]2+ 45 (ppy = 2-phenylpyridine) which was synthesized initially as a mixture of three stereoisomers (meso Δ,Λ; Λ,Λ; Δ,Δ). In this case, all three stereoisomers were separated by preparative thin layer chromatography (SiO2, CH2Cl2/toluene) [92].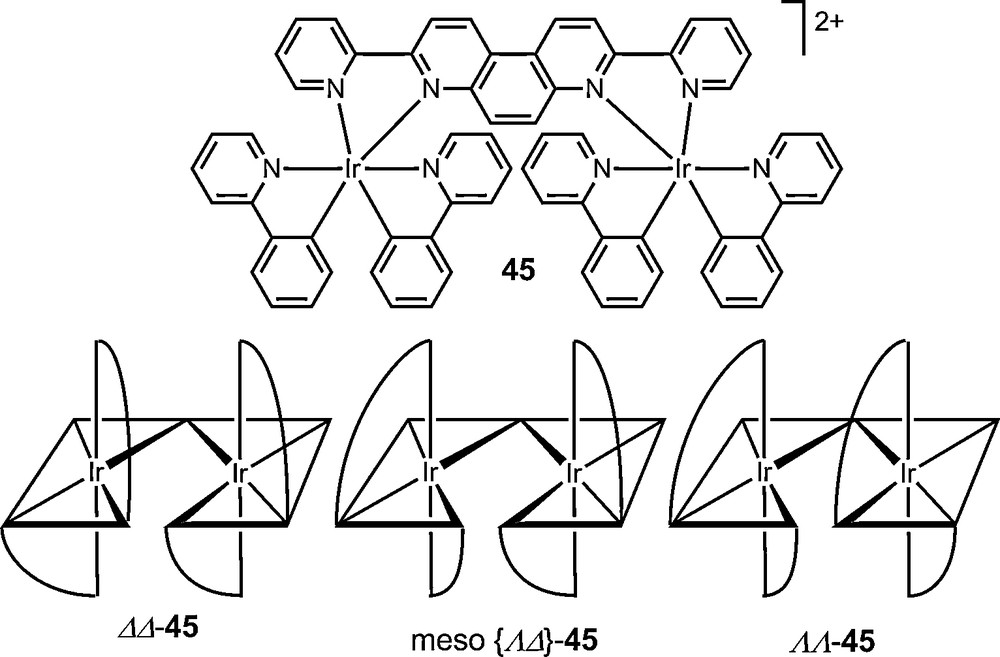
3.3 By asymmetric extraction
The lipophilicity of the TRISPHAT anion 6 also confers to its salts an affinity for organic solvents and, once dissolved, the ion pairs do not partition in aqueous layers. This rather uncommon property can be used to develop a simple and practical resolution procedure of chiral cationic coordination complexes by asymmetric extraction [93,94]. The extraction and the resulting selectivity arise from the preferential binding in the organic phase of one enantiomer of the substrate with the chiral lipophilic selector. It was used recently by Brodie and Aldrich-Wright for the resolution of bromo- and chloro-substituted ruthenium complexes of type 46, precursors to interesting enantiopure bis-intercalating dimers [95].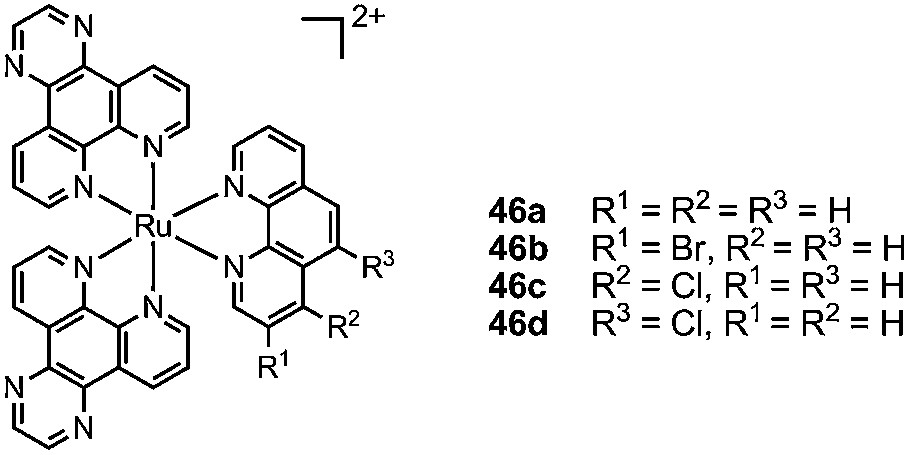
4 Effective asymmetry-inducing agents
Quite a few chiral compounds are configurationally stable as solids and labile in solution. When optically active samples of such derivatives are solubilized, a racemization occurs due to the free interconversion of the enantiomers in solution. If the chiral compounds are charged, an interesting strategy to obtain these molecules in one predominant configuration over time is to consider their asymmetric ion pairing with chiral counter-ions; intermolecular diastereoselective interactions happen and favour one of the equilibrating diastereomers. This so-called Pfeiffer effect [96–100] was evidenced for a series of derivatives [30,53,101,102]. Only a few cases reported over the last few years are now detailed.
In the case of NHC precursor 34 (Fig. 5), it was shown that BINPHAT 11 controls also the configuration of the imidazolium salt with a diastereoselectivity excess up to 27% (CD2Cl2, 213 K) [71]. In the case of the trimesitylmethyl phosphonium cation 35 (Fig. 6), salts 35 [Δ-6] and 35 [Δ-11] were prepared, and the chiral induction (recognition) was studied by 1H and 31P NMR spectroscopy (toluene-d8). Honest diastereomeric excesses were measured (de 39 and 47% respectively, toluene-d8) [72].
Propeller isomerism of trimesitylmethylphosphonium cation 35.
Chiral concave N-heterocyclic carbene 34.
Monomethine dyes 47 were also studied with success in the presence of anions of type 13 (Fig. 2, de up to 77%, C6D6). Copper(I) bis(diimine) complexes 48 and many labile iron(II) tris(diimine) metal complexes were also paired with the chiral counterions and 6 in particular. The NMR signals of the chiral cations were split by the presence of the anions and diastereomeric ratios up to 49:1 were measured for many of the substrates. Some particular studies stand out. First, Constable et al. showed that the configuration of the metal core of ribose-decorated iron(II) metallostar 49 (Fig. 8), is better controlled though interionic diastereomeric interactions with TRISPHAT 6 than by the intrinsic proximity of the chiral sugars [103]. Kol et al. showed that very high chiral induction can be achieved even in polar media despite lower electrostatic attraction and stronger solvent competition via a careful choice of ligands on the iron(II) center, and polycyclic elatin ligand in particular (Fig. 8, complex 50: de > 95% in 90% acetone/chloroform) [104]. Stoddart et al. have used the NMR enantiodifferentiation provided by anion 11 to establish that tetracationic [2]catenane 51 adopts helical conformations in solution (Fig. 9). At low temperature (197K), a preference in favor of either P or M conformation was further noticed in acetone-d6 (2:1 ratio); something remarkable considering the polar nature of the solvent [105]. Recently, chiral [Mo3S4Cl3(dppe)3]+ 52 (dppe = 1,2-bis(diphenylphosphinoethane)) was studied in presence of anion 6. This cluster was shown to be configurationally stable at 20 °C but labile at 72 °C thanks to a modest yet definite stereoinduction (1.2:1) in favor of the heterochiral ion pairs, e.g. [M-52] [Δ-6] [106]. Ditopic ligands incorporating two 2,2’:6’,2”-terpyridine (tpy) ligands linked to a central naphthalenediyl core by ethyleneoxy chains of various lengths were prepared. Their iron(II) complexes yielded in some instances [1 + 1] ferracycles. For such compounds of type 53, the linker between the two tpy domains is spatially restricted and the cations exhibit chirality associated with the conformation about the central N-Fe-N bonds. An effective enantiodifferentiation and decent level of induction was noticed for salt 53 [Δ-11] (de 60% in 2% DMSO-d6/CDCl3). The absolute configuration of the cation in the diastereomeric ion pair was further calculated using TDDFT methods based on ECD measurements [107].
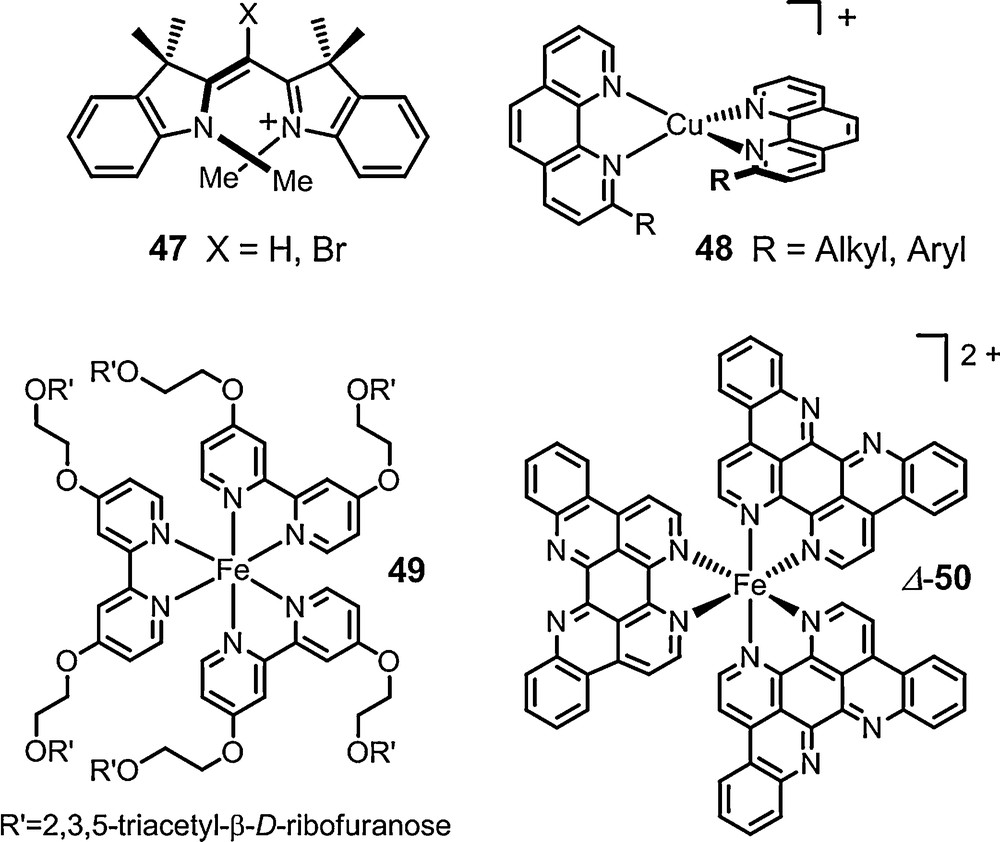
Some configurationally labile cations.
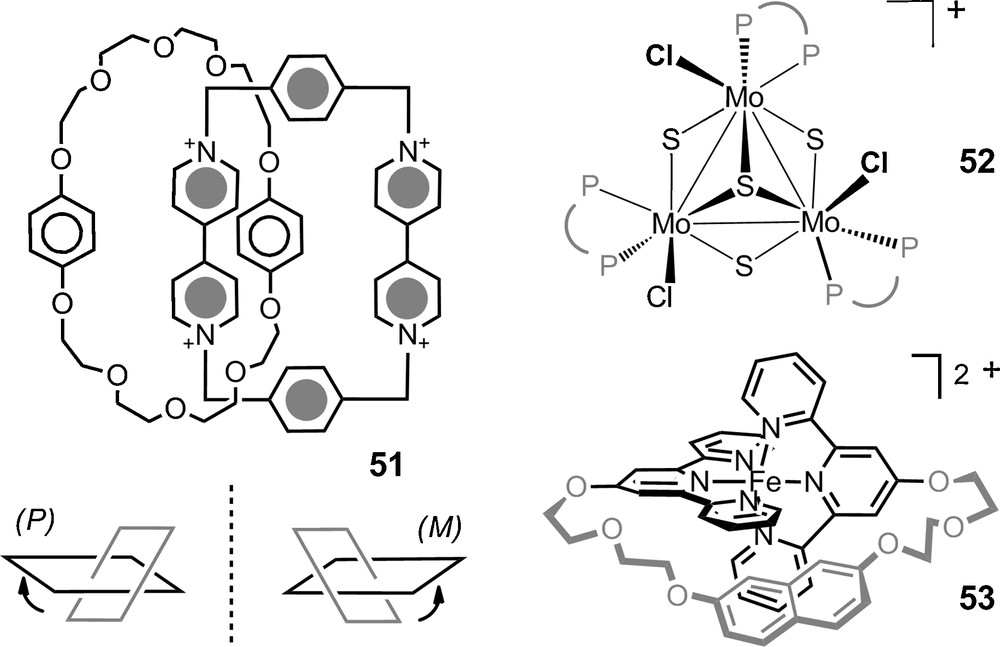
Other configurationally labile cations.
5 Superior ion pairing properties
All the previous examples show the importance of cation–chiral anion interactions and how they can affect the properties of molecules at their proximity. However, the chiral anion can influence the chemical and physical property of the cation itself and a series of reports in the field of supramolecular chemistry have highlighted such effects.
For instance, anion 6 enhances the binding affinity of cationic guests within classical host-guest systems. In the context of quaternary ammonium recognition by cyclophanes or crown-ethers, the presence of 6 as anionic counterion increases the binding affinity of the ammonium salts by 2 to 7-fold. Currently, it seems that 6 is the best counterion ever reported for ammonium host-guest chemistry. It was used to demonstrate that full ion pairs, and not just the ammonium cation, must be considered in host-guest studies in CHCl3 leading to interesting photophysical properties [108] or, in conjunction with chiral cyclic hexapeptidic hosts of type 54, to effective matched/mismatched asymmetric ion pairing situations [109,110].
This knowledge was used further in a study directed towards the stereoselective synthesis of inherently chiral pseudorotaxanes 55. 1H NMR spectra (parts, Fig. 10) of dissymmetric aryl-substituted macrocycle DB24C8F6 are reported for the PF6− (left) and rac-TRISPHAT (right) salts; U and T indicating the unthreaded and threaded (diastereomeric) macrocycle respectively. Clearly, a much higher concentration of diastereomeric pseudo-rotaxanes is observed in the presence of the lipophilic counterion 6; the threading process with TRISPHAT counterions being thus much more effective (× 7.3) than with the PF6− analogue [111].
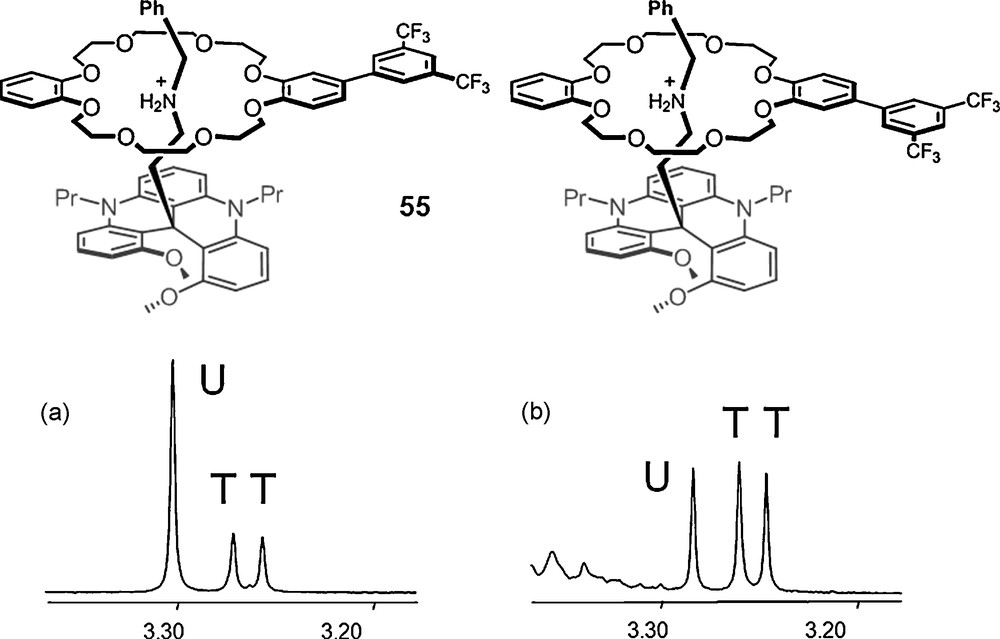
1H NMR spectra (500 MHz, parts, CDCl3) of dissymmetric aryl-substituted macrocycle DB24C8F6 – U and T indicate the unthreaded and threaded (diastereomeric) macrocycle respectively: (a) PF6− salt; (b) rac-6 salt.
In a somewhat related study, it was shown by Laursen et al. that TRISPHAT anions 6 can have a profound influence upon the ratio of translational isomers in bistable donor-acceptor rotaxanes where a p-accepting tetracationic cyclophane shuttles between two different p-donating recognition sites – the presence of anions 6 as counterions favoring predominantly the encirclement of bispyrrolotetrathiafulvalene over dioxynaphthalene recognition sites [112].
Finally, most of the above-described examples have used the chiral anions in enantiomerically enriched or pure forms. Applications of the chiral anions in their racemic form are also possible. Maury, Le Bozec, Ledoux and coworkers have used the lipophilicity of racemic TRISPHAT to enhance the solubility of cationic, octupolar nonlinear optically active, metal trisbipyridyl complexes, and their polymeric or dendrimeric forms in particular [113–116]. Salts of various racemic cations and anions 6 crystallize readily in centrosymmetric unit cells providing interesting new phases or supramolecular arrangements as a result [117–119]. In terms of synthetic applications, the superior lipophilicity that TRISPHAT anions confers to its salts allows the perfect containment in organic layers of reactive cationic systems when reactions are performed in biphasic CH2Cl2/water conditions. This effective partitioning of the organic reagents was recently used for the development of enantioselective olefin epoxidation reactions mediated by catalytic iminium/oxaziridinium species; compounds 56 and 57 being typical examples of the iminium catalysts used (Fig. 11) [120–122]. The strict biphasic CH2Cl2/water conditions that can be enforced due to the presence of anions 6 can enhance the selectivity of the epoxidation reaction and help the recovery of the non-racemic epoxides.
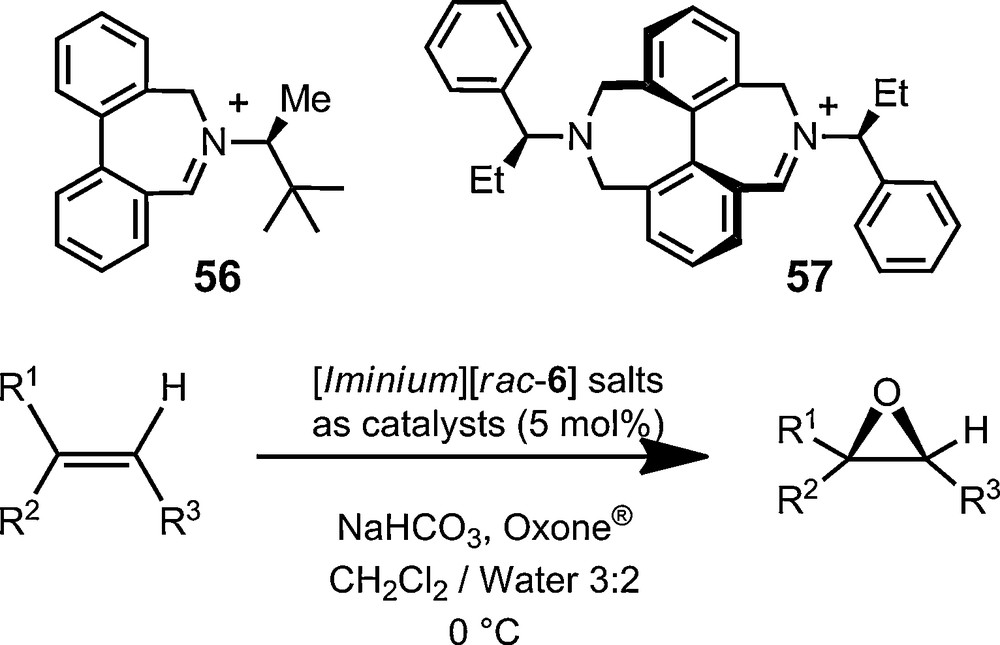
Biphasic enantioselective epoxidation reactions mediated by the lipophilic TRISPHAT anion 6.
6 Enantioselective synthesis and catalysis
Until recently, chiral anionic counterions were essentially unused by the synthetic organic community as these species had displayed only minute influence on the stereochemical outcome of reactions involving cationic intermediates or reagents. This has changed rather dramatically over the last three years with a series of elegant reports displaying high selectivity [13]. In these papers, effective anions have all been tetracoordinated BINOL phosphate anions rather than six-coordinated derivatives.
The only exception was the recent report that an enantioselective [1,2]-Stevens rearrangement is feasible using such a chiral anion strategy. Diphenylazepinium cations of type 58 were associated with BINPHAT 11. Upon association, thanks to a Pfeiffer effect, a partial stereocontrol of the axial chirality of the cations occurs (Fig. 12, de 20–60% as a function of substituent Z). This chiral induction is then effectively transferred to the centered chirality of rearranged amines 59 upon treatment of salts [57,11] with a strong neutral Schwesinger's base (ee up to 55%, transfer of chirality 90 to 100%) [123].
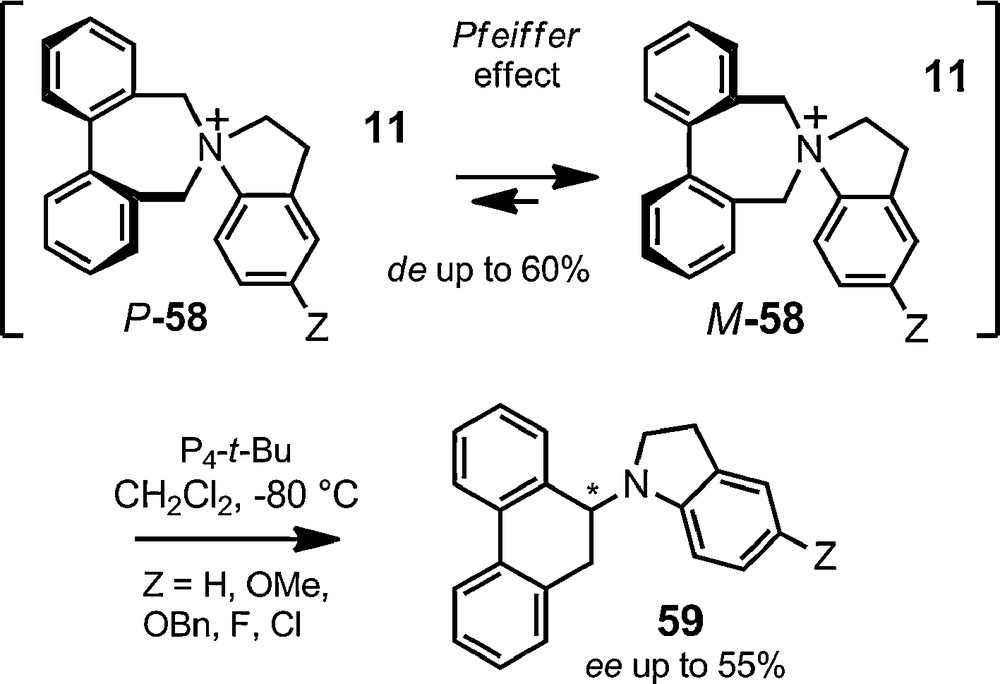
Enantioselective [1,2]-Stevens rearrangement.
7 Conclusion
In conclusion, hexacoordinated phosphate anions like TRISPHAT 6 and BINPHAT 11 are chemically and configurationally stable anions which behave as able NMR solvating, resolving and asymmetry-inducing reagents when paired with configurationally stable or labile chiral cations. The organic, inorganic or organometallic nature of the cationic moieties is not influential as good anionic matches can usually be found. We believe that there is much to gain from this supramolecular approach to stereoselective synthesis as both configurations of a chiral cationic complex can be generated in high diastereomerical purity with no need to prepare two sets of enantiomeric ligands. It is a priori sufficient to form the cationic derivative with achiral ligands and exchange the traditional achiral anions (PF6−, BF4−, etc.) for chiral versions.
Acknowledgements
We are grateful for financial support of this work by the University of Geneva, the Swiss National Science Foundation and the State Secretariat for Research and Science.
1 To limit the scope of the review, only examples of asymmetric chemistry involving “unreactive” chiral anions will be presented. Chiral organolithiums or Grignard reagents, which can be considered as chiral anions, will therefore not be covered in this article. For a recent review on this topic, see [11].
2 pKa for first ionization of catechols can be used as a measure of the electronic effect of the substituents on a catechol; tetrachlorocatechol: pKa1 = 6.63; pyrocatechol: pKa1 = 10.87.
3 For a chiral complex with the ligand in C2-symmetric conformation, all atoms are chirotopic, and a doubling of all 1H and 13C NMR signals would have resulted.