

1 Introduction
Organic π-conjugated materials are an important class of compounds due to their significant potential for diverse practical applications in optoelectronics, including light-emitting diodes, field-effect transistors, and photovoltaic cells [1]. Their organic nature provides for a plethora of synthetic possibilities to fine tune the materials’ properties toward specific applications (addressing, e.g., semiconducting behavior, or processability). In recent years, the use of organophosphorus components (as well as boron and silicon compounds) has become a promising new approach in this context, as the presence of the phosphorus center(s) introduces unique electronic features to π-conjugated materials, which are not accessible in this simplicity with native organic components [2]. Over the past seven years or so, we were able to comprehensively illustrate the beneficial features accessible through organophosphorus electronics using the highly luminescent dithieno[3,2-b:2′,3′-d]phosphole system [3]. Through our systematic structure-property studies, we were able to address a variety of important aspects including tunability of the photophysical properties, solid-state organization, processability (i.e., through polymeric architectures), as well as the redox-behavior of the materials [4]. However, synthetic methods that easily afford mono-substituted, or asymmetric, oxidized dithieno[3,2-b:2′,3′-d]phospholes are still rare [5].
In this contribution, we now report on the synthesis and characterization of acylated dithieno[3,2-b:2′,3′-d]phosphole oxides prepared via Friedel-Crafts acylation. Interestingly, this reaction provides a facile synthetic route to asymmetric, mono-functionalized π-conjugated phosphole-based materials. These can be further postfunctionalizated through bromination of the vacant 6-position of the dithienophosphole scaffold and homo-coupled under Yamamoto reaction conditions, demonstrating that extended π-conjugated oligomers with further tuned absorption and emission properties can be realized.
2 Results and discussion
2.1 Synthesis
Friedel-Crafts acylation of dithienophosphole oxides has successfully been employed for the preparation of symmetric and asymmetric luminescent materials. As shown in Scheme 1, the synthetic route utilized the treatment of the dithieno[3,2-b:2′,3′-d]phosphole oxide 1 with the appropriate acyl chlorides in the presence of excess AlCl3 under mild reaction conditions in dichloromethane to afford compounds 2, and 3. Surprisingly, even an excess of the acyl chloride (∼3.0 equivalents), yielded only mono-functionalized phospholes, with the exception of the formation of a minor amount of the 2,6-difunctionalized phosphole (< 5%), when the acylation reagent was acetyl chloride, denoted as compound 2x. The disubstituted species 2x could be selectively crystallized from an ethyl acetate/hexanes solution at room temperature. Reactions employing thiophene-2-carbonyl chloride, on the other hand, did not form the corresponding difunctionalized phosphole, even under extended reaction times (> 2 days). The identity of the mono-functionalized species was supported by the heteronuclear NMR studies.
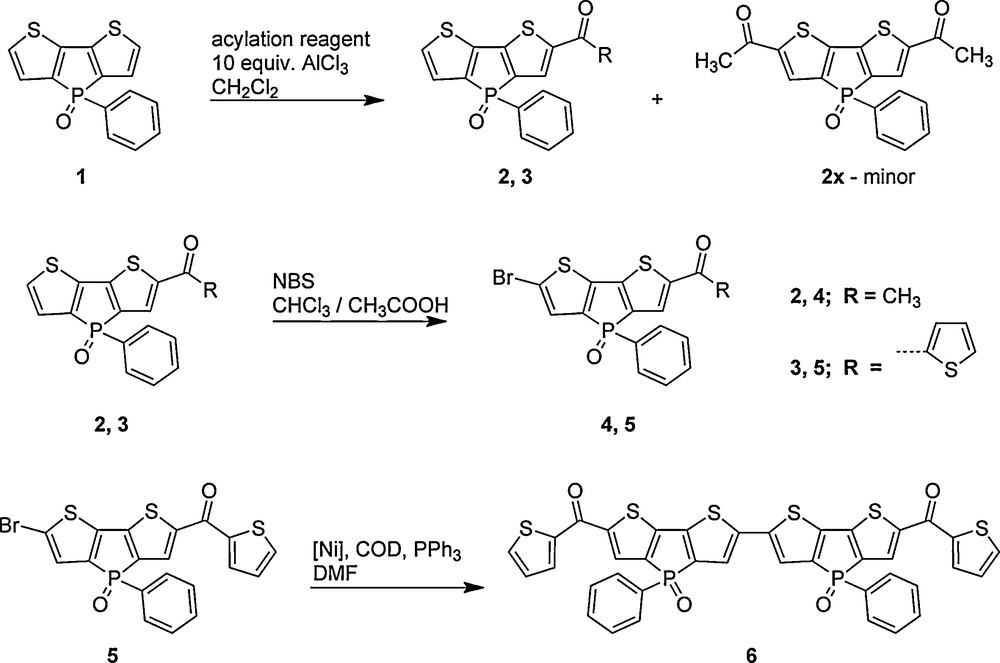
Synthetic route to asymmetric and symmetric carbonyl-substituted dithieno[3,2-b:2′,3′-d]phospholes, compounds 2 through 5, and compound 6, respectively.
Subsequent treatment of 2 and 3 with N-bromosuccinimide (NBS) in acetic acid/chloroform yielded mono-brominated compounds 4 and 5 in good yields after a short silica column using ethyl acetate as an eluent or crystallization from acetone, respectively. In order to demonstrate that the mono-brominated acylated phospholes could also be used to yield materials with more extended conjugation, nickel-catalyzed Yamamoto coupling [6] was employed using 5 as a representative example (Scheme 1). The resulting compound 6, was found to possess the desired red-shifted absorption and emission properties (vide infra), however, exhibited limited solubility in common organic solvents making more extensive optical and electronic characterizations difficult.
2.2 Crystal structures
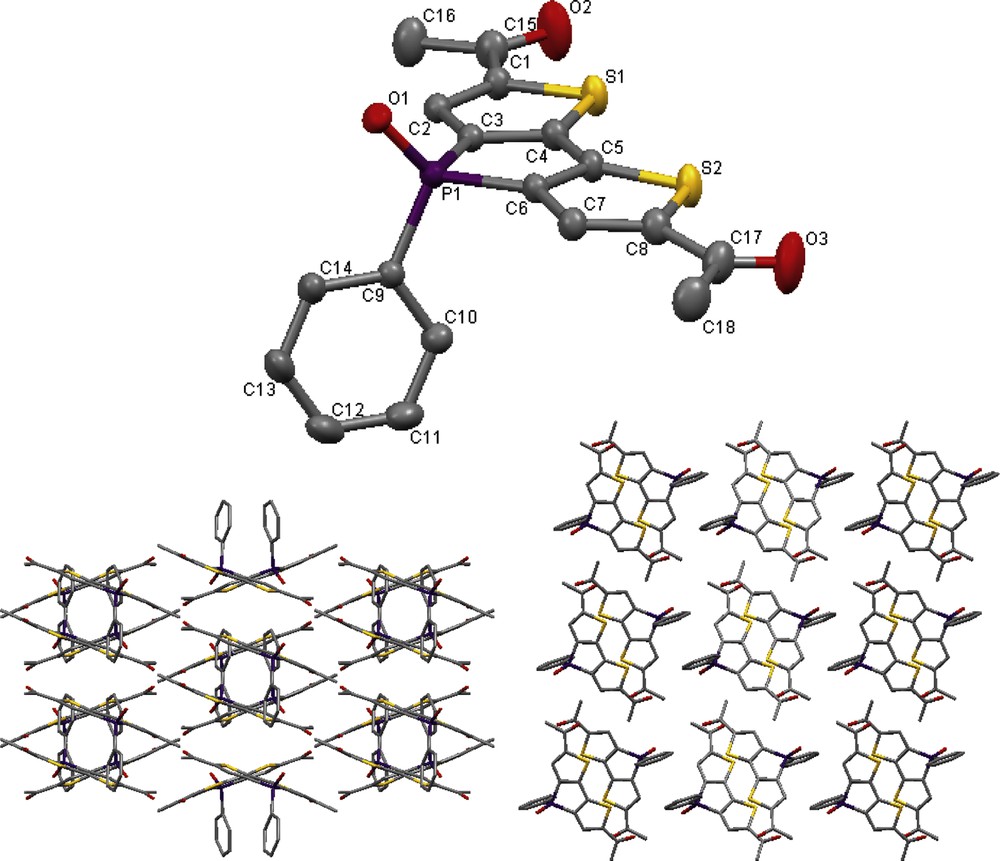
As stated in section 2.1, a minor quantity of the diacetyl product (2x) was isolated through crystallization from a concentrated ethyl acetate/hexanes solution and suitable for single crystal X-ray structure analysis. The molecular structure and crystal packing of 2x are shown in Fig. 1. Suitable single crystals of 3 were obtained from a concentrated ethanol solution, and its molecular structure and packing are shown in Fig. 2. Table 1 summarizes selected bond lengths for compounds 2x and 3.
Molecular structure (top) and packing (bottom) of 2x in the solid state (50% probability level, H atoms omitted for clarity).

Molecular structure (left) and packing (right) of 3 in the solid state (50% probability level, H atoms omitted for clarity).
Selected bond lengths for compounds 2x and 3.
| 2x | 3 | ||
| (O1 – P1) | 1.483(1) Å | (O1 – P1) | 1.483(7) Å |
| (C3 – P1) | 1.820(5) Å | (C3 – P1) | 1.810(2) Å |
| (C9 – P1) | 1.789(2) Å | (C6 – P1) | 1.806(2) Å |
| (C1 – C2) | 1.377(3) Å | (C14 – P1) | 1.802(2) Å |
| (C1 – C15) | 1.472(3) Å | (C4 – C5) | 1.456(3) Å |
| (C4 – C5) | 1.460(3) Å | (C5 – C6) | 1.384(4) Å |
| (C15 – O2) | 1.218(3) Å | (C8 – C9) | 1.467(3) Å |
| (C15 – C16) | 1.499(3) Å | (C9 – O2) | 1.228(3) Å |
| (C9 – C10) | 1.472(3) Å |
The molecular structures of 2x and 3 exhibit similar characteristics with respect to the planarity of the conjugated backbone with the exception that the carbonyl-thiophene in 3 twists slightly out of the plane by approximately 4°, whereas the respective orientation of the pendant P-phenyl moiety is rotated approximately 90° relative to 2x, likely due to packing effects. Both compounds also exhibit similar orientations of the carbonyl moieties, lying in the same plane as the thiophene S atoms, similar to a diformyl-functionalized phosphole reported earlier [7]. The thiophene carbonyl bond lengths and twisting are similar to those found in a related di-2-thienyl ketone [8]. However, the molecules of 2x show little direct overlap in the solid-state presumably due to the bulkier terminal methyl moieties. Interestingly, mono-substitution of 3 imparts chirality about the P atom and both enantiomers are observed interacting in a slip-stacked pair packing arrangement as shown in Fig. 2.
2.3 UV-vis and fluorescence spectroscopy
Solution absorption and fluorescence data for compounds 1 through 6 are summarized in Table 2 and the absorbance and photoluminescence spectra are depicted in Fig. 3. As shown by the black plots of compounds 1, 2 and 3 (Fig. 3), the absorption maxima (λmax) exhibit a bathochromic shift with the addition of acetyl (2; λmax = 381 nm) and thiophene-2-carbonyl (3; λmax = 393 nm), relative to unsubstituted dithienophosphole (1; λmax = 361 nm, [3]). The red-shift is attributed to an extension of the π-system with the addition of the ketone moiety. The brominated compounds 4 and 5 exhibit a further red-shift in absorbance, as compared to the acylated derivatives, shifting by Δλmax = 13 and Δλmax = 11 nm, respectively. Extinction coefficients (ɛ) were extracted from λmax for compounds 1, 2 and 3 (Table 2). The unsubstituted dithienophosphole 1 exhibits the lowest extinction coefficient at ɛ = 6250 M−1cm−1, relative to the acylated and brominated compounds 2 through 5. The value for compounds 2 (ɛ = 12,444 M−1 cm−1) nearly doubles to ɛ(4) = 24,444 M−1 cm−1 upon bromination. Compounds 3 and 5 showed similar absorption characteristics increasing the extinction via ɛ(3) = 14,909 M−1 cm−1 after acylation, to ɛ(5) = 23,880 M−1 cm−1 post-bromination. Surprisingly, the ɛ of compound 5 is slightly lower than compound 4 possibly indicating that the thiophene moiety is not completely coplanar with the phosphole unit. Yamamoto coupling of 5 increases the extinction coefficient to a greater extent, nearly tripling, increasing to ɛ(6) = 60,000 M−1 cm−1 (λmax = 438 nm). The increase, relative to the unsubstituted phosphole and brominated compound, is attributed to the extended absorption cross-section of the molecule and extended π-system. Density Functional Theory (DFT) calculations, at the B3LYP/6-31G(d) level of theory [9], indicate that the lone pair(s) at the Br atom contribute to the π– and π*- system, thereby extending the frontier orbitals (HOMO, LUMO) and the absorption cross-section.
Photophysical data of compounds 1– 6 in dichloromethane solution.
| Compound | λmax/nm | ɛ/M−1 cm−1 | λem/nm | ϕPL | η/ns |
| 1 | 361 | 6250 | 447 | 0.21 | 13 |
| 2 | 381 | 12,444 | 463 | 0.26 | 6 |
| 3 | 394 | 24,444 | 478 | 0.11 | 1 |
| 4 | 393 | 14,909 | 469 | 0.46 | 6 |
| 5 | 404 | 23,880 | 483 | 0.30 | 2 |
| 6 | 438 (520) | 60,000 | 560 (605) | 0.31 | NA |

UV-vis absorption and photoluminescence spectra of compounds 1 a) and b), 2 c) and d), and 3 e) and f) in dichloromethane solution.
Photoluminescence spectra for compounds 1, 2, and 3 also exhibit red-shifted characteristics, shifting from λem = 447 nm (unsubstituted 1, [3]) to λem = 463 and λem = 469 nm for the non-brominated compounds 2 and 3, and λem = 478 and λem = 483 nm for the brominated compounds 4 and 5, respectively. Photoluminescence quantum yields (ϕPL) were obtained for dichloromethane solutions of 1, 2, and 3; however, most values range between ϕPL = 11–31%, which is significantly lower than usually observed for dithienophospholes (ϕPL = 35–90%) [3,4]. The lower values can be attributed to the quenching character of the incorporated ketone functionalities, opening up significant non-radiative relaxation pathways, and are in line with earlier observations for a diformyl-functionalized dithienophosphole [7]. Surprisingly, after bromination of compounds 2 and 3, the quantum yield was found to increase. DFT-calculated molecular orbitals of compound 4 (not shown) support that bromine is an integral component of the π– and π*– system. However, the calculations also show that bromination only effects the energy level of the LUMO by lowering it to −2.58 eV (4) from −2.44 eV (2), whereas the energy of the HOMO remains essentially unchanged at −6.05 eV (cf. 2: −6.04 eV). This suggests that bromine acts as an electron acceptor. Although a rare phenomenon, Havlas and Michl have recently reported that the heavy atom effects of bromine can indeed be inverted when the bromine behaves as an electron acceptor [10]. Thus, it is plausible that a similar mechanism may apply in the present study. Photoluminescence life times (η) were also measured for compounds 1 through 5, and show typical values for organophosphorus-based conjugated materials (Table 2) [11].
During the synthesis of 2 and 3, the emission color of the reaction mixtures shifted from blue to yellow upon addition of AlCl3 leading us to explore whether it was possible to further tune the emission color of the resulting phospholes in the presence of Lewis acids. Therefore, the photophysical properties of compounds 1, 2 and 3 in the presence of BF3 were investigated to examine the spectral response, which was believed to originate from a Lewis-acid complex to the oxygen atoms of the PO and/or CO functional groups of the scaffold. As shown in the red and blue plots of Fig. 3a, c, and e, compounds 1 and 2 exhibited a red-shifted absorbance profile upon addition of the Lewis-acid. The shifts in λmax for compounds 1 and 2 were Δλmax = 7 and Δλmax = 4 nm for the initial addition of Lewis-acid and Δλmax = 15 and Δλmax = 7 nm after the addition of a large excess of boron trifluoride (employed as diethyl etherate), relative to the position of λmax of the neat solutions, respectively. It should be noted that the initial addition of boron trifluoride was chosen to be approximately 10 times that of the phosphole concentration in order to observe any spectral shift more easily, as adding one equivalent per carbonyl moiety did not perturb the spectral properties in a measurable way. Interestingly, compound 3 did not exhibit a shift in absorbance even with the addition of a large excess of boron trifluoride. An interesting trend that is observed for compounds 1, 2 and 3, after the addition of boron trifluoride, is the red-shift in λmax, as compared with the neat solutions, decreases as the extent of conjugation of the molecules increases.
Contrary to the absorption spectroscopy results, all of the photoluminescence spectra for compounds 1, 2 and 3 exhibited red-shifted emission characteristics upon addition of boron trifluoride. Compound 1 exhibited the strongest red-shift in emission, shifting from λem = 447 nm to λem = 490 nm, whereas compounds 2 and 3, exhibited nearly similar shifts, shifting from λem = 463 to λem = 493 nm and λem = 469 to λem = 495 nm, respectively.
31P and 13C NMR spectroscopy experiments were employed to investigate the coordination of BF3 to the PO and/or CO moieties in chloroform solutions of compounds 1 and 3. Both compounds exhibited a downfield shift of approximately Δδ = 20 ppm in their 31P NMR spectra after the addition of BF3. On the contrary, the 13C NMR resonance peak associated with the carbonyl moiety, located at ∼177 ppm, did not exhibit any shift upon addition of BF3. These results suggest that the interaction of the Lewis acid, i.e., BF3, with the dithienophospholes prepared in this study is mostly associated with the phosphorus-oxygen moiety. The observed optical responses for compounds 1–3 suggest that the effect of the functionalized phosphorus center on the photophysical properties of the materials as a whole diminishes upon increasing extent of π-conjugation, which is supported by the frontier orbitals of the three compounds (vide infra). This feature has also recently been observed with extended donor/acceptor substituted dithienophospholes that do not show any shift of the photophysical data upon significant modification of the phosphorus center (oxidation or complexation to Au) [12].
The observations confirm that the photophysical properties of acylated and non-acylated dithienophospholes can be further tuned through addition of Lewis acids and the effects on the absorption and/or emission features depend on the degree of conjugation within the (extended) dithienophosphole scaffolds. A similar behaviour was recently also reported for somewhat related, 2,1,3-benzothiadiazole-functionalized extended dithienosiloles [13].
2.4 Theoretical calculations
To explore the extent of delocalization in the molecular orbitals of the acylated dithienophospholes, suggested by the bathochromic spectroscopic shifts in the absorption and emission spectra, DFT was employed at the B3LYP/6-31G(d) level [9]. The resulting isodensity plots are shown in Fig. 4 for compounds 1, 2 and 3. All of the compounds display a similar distribution in their respective highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) and extending over the carbonyl moiety for compounds 2 and 3. The HOMO level of compounds 2 and 3 are nearly degenerate (2: EHOMO = −6.04 eV, 3: EHOMO = −6.01 eV) and lower in energy than unsubstituted dithienophosphole 1 (cf.: EHOMO = −5.77 eV). The calculated energy levels of the LUMOs are also markedly different from compound 1, lowering in energy from ELUMO = −1.86 eV (1) to ELUMO = −2.44 and −2.52 eV for compounds 2 and 3, respectively. The difference in ELUMO for 2 and 3 is due to the more extended π-system with the addition of the thiophene moiety of 3 (Fig. 4). The trend in the energy gaps (Eg) of compounds 1, 2 and 3, i.e., Eg = 3.91, 3.60 and 3.50 eV, respectively, correlates well with the decreasing trend observed in the optical energy gap (Eopt) extracted from the λmax of absorption, i.e., Eopt = 3.44, 3.26 and 3.16 eV, respectively, possibly due to the increasing extent of conjugation [1]. The relative values differ likely due to the level of the basis set employed, as well as the fact that the molecules were not modeled in solvent.

Isodensity plots for compounds 1, 2 and 3 and their associated HOMO and LUMO energy levels calculated at the B3LYP/6-31G(d) level of theory.
3 Conclusions
In conclusion, we have demonstrated that Friedel-Crafts acylation conditions provide an easy and efficient way to synthesize 2-mono-functionalized dithienophospholes, which are otherwise very difficult to obtain. Acylation of the main scaffold was found to provide extended π-conjugated systems with red-shifted photophysical properties, when compared to the dithienophosphole core. Acylation further induced a significant increase in the absorption features, and also allows for additional tuning of the photophysical properties through interaction with Lewis acids, likely involving the PO group. The mono-acylated species can further be functionalized with a bromo-substituent in the 6-position that permits subsequent cross-coupling reactions to afford bis(dithienophosphole) oligomers with strongly red-shifted photophysical properties and an enhanced absorption profile. It is believed that the addition of alkylated thiophene-2-carbonyl chlorides to the phosphole would yield more soluble derivatives that may possess interesting liquid crystalline properties and will be discussed in a future publication.
4 Experimental
All the reactions were carried out under nitrogen atmosphere using standard Schlenk techniques. Unless otherwise specified, reagents were used as received. The detailed preparation of compound 1 has been reported elsewhere [3]. 1H NMR, 13C{1H} NMR, and 31P{1H} NMR spectra were recorded on Bruker DRY-400, and Bruker UGI-400 spectrometers. Mass spectrometry was performed using a Finnigan SSQ700 or Bruker Daltonics AutoFlex III system. Optical spectroscopy experiments were recorded in dichloromethane solution using a UV-vis-NIR Cary 5000 spectrophotometer and Edinburgh Instruments Ltd FLS920P fluorescence spectrometer equipped with an integrating sphere. Solutions were degassed with N2 prior to measuring photoluminescence. Lewis-acid complexation studies were performed by the initial addition of 2.5 × 10−7 mol boron trifluoride diethyl etherate to a solution of compounds 1, 2, or 3 in degassed dichloromethane solution followed by the addition of a large excess of boron trifluoride diethyl etherate (1.27 × 10−4 mol) to ensure full complexation of the Lewis-acid to the phosphole. Additional aliquots of boron trifluoride diethyl etherate were added; following the excess addition, however, changes in absorption or photoluminescence were not observed. Diffraction data was collected on a Nonius Kappa CCD diffractometer using monochromated Mo Kα radiation (λ = 0.71073 Å). The datasets were corrected for Lorentz and polarization effects and absorption correction was applied to the net intensities (Table 3). The structure was solved by direct methods and refined using SHELX [14]. After full-matrix least-square refinement of the nonhydrogen atoms with anisotropic thermal parameters, the hydrogen atoms were placed in calculated positions using a riding model.
Crystal data and structure refinement for 2x and 3.
| 2x | 3 | |
| Formula | C18 H13 O3 P S2 | C19 H11 O2 P S3 |
| Mr | 372.37 | 398.43 |
| T | 173(2) K | 123(2) K |
| λ | 0.7107 Å | 1.54178 Å |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P21/c |
| a | 20.20720(70) Å | 8.8545(3) Å |
| b | 8.24900(29) Å | 11.2803(3) Å |
| c | 20.88330(50) Å | 17.7894(5) Å |
| α | 90° | 90° |
| β | 97.1208(17)° | 106.6930(10)° |
| γ | 90° | 90° |
| V | 3454.171(192) Å3 | 1701.95(9) Å3 |
| Z | 8 | 4 |
| ρcalcd | 1.432 Mg/m3 | 1.555 Mg/m3 |
| μ | 0.414 mm−1 | 4.962 mm−1 |
| F(000) | 1536 | 816 |
| Crystal size | 0.16 × 0.09 × 0.06 mm3 | 1.178 × 0.37 × 0.366 mm3 |
| θ range | 1.97 to 27.46° | 4.7 to 68° |
| Index ranges | −25 ≤ h ≤ 26 | −8 ≤ h ≤ 10 |
| −10 ≤ k ≤ 10 | −13 ≤ k ≤ 12 | |
| −26 ≤ K ≤ 26 | −18 ≤ K ≤ 20 | |
| Reflections collected | 7185 | 10559 |
| Independent reflections | 3883 [R(int) = 0.0216] | 3029 [R(int) = 0.0489] |
| Completeness to θ | 98 % | 97.8 % |
| Absorption correction | Semi-empirical | Numerical |
| Max./min. transmission | 0.9756 and 0.9367 | 0.4376 and 0.1039 |
| Data/restraints/parameters | 3883/0/225 | 3029/0/226 |
| GoF on F2 | 1.225 | 1.076 |
| Final R indices [I > 2σ(I)] | R1 = 0.0436, | R1 = 0.0448, |
| R indices (all data) | R1 = 0.0488, | R1 = 0.0453, |
| Largest diff. peak/hole | 0.524 and −0.514 eÅ−3 | 0.657 and −0.511 eÅ−3 |
4.1 Compounds (2) and (3) were prepared using a similar procedure
1.72 g (11.7 mmol) of AlCl3 was dissolved in dry DCM. 0.390 g (2.67 mmol) of thiophene-2-carbonyl chloride (in the preparation of 3) was added into the flask. 0.310 g (1.07 mmol) of the phosphole oxide 1 was dissolved in dry DCM and added into the flask containing the AlCl3 and acyl chloride. The reaction was stirred overnight at room temperature. The fluorescence changed from yellow-green to yellow-orange during the course of the reaction. The solution was quenched over sodium bicarbonate water and the organic layer separated. The compounds were extracted from the aqueous layer with DCM three times, followed by washing with aqueous sodium bicarbonate, water, and lastly saturated NaCl. The organic layer was dried with MgSO4, filtered, and evaporated yielding a yellow powder (2) or orange oil (3). Compound 2 was further purified on a silica column using ethyl acetate as an eluent followed by recrystallization from acetone. Analysis of column fractions yielded a minor amount of the di-reacted product 2x for which the crystal structure was determined. Compound 3 was further purified on a silica column with 4:1 ethyl acetate:hexane as the eluent and recrystallized from ethanol yielding yellow crystals.
(2) Yield: 0.359 g, 1.13 mmol, 71.6%; 31P-NMR (CD2Cl2, 162 MHz): δ = 17.88 (s, 1P) ppm; 1H-NMR (CD2Cl2, 400 MHz): δ = 7.74–7.68 (m br, 3H), 7.60–7.55 (m br, 1H), 7.52–7.50 (dd, 1H, J = 4.8, J = 3.2 Hz), 7.50–7.43 (m br, 2H), 7.22–7.20 (dd, 1H, J = 4.8, J = 2.4 Hz), 2.50 (s, 3H) ppm; 13C-NMR (CD2Cl3, 100 MHz): δ = 190.73 (s), 148.50 (d, J = 11.6 Hz), 145.59 (d, J = 23.1 Hz), 142.61 (d, J = 109.6 Hz), 140.91 (d, J = 109.2 Hz), 139.38 (s), 133.30 (d, J = 2.9 Hz), 132.16 (d, J = 14.8 Hz), 131.40 (d, J = 11.1 Hz), 131.19 (d, J = 13.2 Hz), 130.51 (d, J = 108.1 Hz), 129.64 (d, J = 13.0 Hz), 126.82 (d, J = 14.0 Hz), 26.88 (s) ppm; HR-MS (TOF MS EI+): calculated for C16H11O2PS2 m/z: 329.9938 [M+], found m/z: 329.9948, mp: 192 – 195 °C.
(2x) 31P-NMR (CDCl3, 162 MHz): δ = 18.10 (s, 1P) ppm; 1H-NMR (CDCl3, 400 MHz): δ = 7.76–7.71 (m br, 2H), 7.69 (d, 2H, J = 2.8 Hz), 7.63–7.58 (dtd, 1H, J = 7.2, J = 1.6 Hz), 7.51–7.48 (m br, 2H), 2.54 (s, 6H) ppm; MS (EI): m/z (%): 371.88 (100) [M+], 356.88 (57) [M+ – CH3], 294.93 (20) [M+ – C6H5].
(3) Yield: 0.510 g, 1.27 mol, 83%; 31P-NMR (CDCl3, 162 MHz): δ = 18.81 (s, 1P) ppm; 1H-NMR (CDCl3, 400 MHz): δ = 7.90–7.89 (d, 1H, J = 2.8 Hz), 7.87–7.86 (dd, 1H, J = 3.6, J = 1.2 Hz), 7.77–7.70 (m br, 3H), 7.59–7.54 (m br, 1H), 7.49–7.43 (m br, 3H), 7.24–7.22 (dd, 1H, J = 4.8, J = 2.4 Hz), 7.19–7.17 (dd, 1H, J = 4.8, J = 3.6 Hz) ppm; 13C-NMR (CDCl3, 100 MHz): δ = 177.94 (s), 152.14 (d, J = 22.1 Hz), 146.64 (d, J = 12.3 Hz), 144.95 (d, J = 23.0 Hz), 141.74 (d, J = 11.1 Hz), 140.52 (d, J = 53.5 Hz), 138.89 (s), 133.95 (d, J = 50.7 Hz), 132.86 (d, J = 2.9 Hz), 131.52 (d, J = 14.9 Hz), 131.19 (d, J = 13.8 Hz), 130.85 (d, J = 11.3 Hz), 129.14 (d, J = 13.2 Hz), 129.06 (s), 128.14 (s), 127.97 (s), 126.47 (d, J = 14.3) ppm; MS (EI): m/z (%): 397.69 (100) [M+], 320.70 (46) [M+ – C6H5], 110.87 (35) [M+ – C14H8S2PO]; HR-MS (TOF MS CI+): calculated for C19H11O2S3P m/z: 397.9659 [M+], found m/z: 397.9641, mp: 210 – 211°.
4.2 Compounds 4 and 5
In a typical bromination reaction, 0.19 g (0.47 mmol) of the keto-phosphole oxide, 2 or 3, was dissolved in chloroform and acetic acid (1:2) mixture. N-bromosuccinimide, 0.092 g (0.51 mmol), was added and stirred for 24 h. The solution was then quenched with NaOH solution and extracted with CHCl3 (3 × 25 mL) and subsequently washed with (3 × 25 mL) with aqueous NaOH. The combined organic layers were dried with MgSO4, filtered and reduced to a yellow powder.
(4) Yield: 0.289 g, 0.710 mmol, 64.7%; 31P-NMR (CD2Cl2, 162 MHz): δ = 18.67 (s, 1P) ppm; 1H-NMR (CD2Cl2, 400 MHz): δ = 7.74–7.68 (m br, 3H), 7.62–7.58 (tdt, 1H, J = 7.2, J = 1.8 Hz), 7.50–7.45 (m br, 2H), 7.23–7.22 (d, 1H, J = 2.4 Hz), 2.50 (s, 3H) ppm; 13C-NMR (CD2Cl2, 100 MHz): δ = 190.67 (s), 151.86 (s), 148.78 (d, J = 8.5 Hz), 145.61 (d, J = 22.9 Hz), 141.82 (d, J = 106.8 Hz), 139.14 (d, J = 110 Hz), 133.55 (d, J = 3.1 Hz), 131.40 (d, J = 11.3 Hz), 131.07 (d, J = 13.6 Hz), 129.82 (d, J = 108.6 Hz), 129.75 (d, J = 13.1 Hz), 129.34 (d, J = 13.7 Hz), 26.90 (s); HR-MS (TOF MS EI+): calculated for C16H10O2PS2Br m/z: 409.9023 [M+], found m/z: 409.9029, mp: 212 – 215 °C.
(5) Yield: 0.19 g, 0.34 mmol, 85%; 31P-NMR (CDCl3, 162 MHz): δ = 19.06 (s, 1P) ppm; 1H-NMR (CDCl3, 400 MHz): δ = 7.89–7.88 (d, 1H, J = 2.8 Hz), 7.86–7.85 (dd, 1H, J = 3.6, J = 0.8 Hz), 7.76–7.70 (m br, 3H), 7.61–7.56 (tdt, 1H, J = 3.2, J = 1.6 Hz), 7.49–7.44 (m br, 2H), 7.20–7.17 (m br, 2H) ppm; 13C-NMR (CDCl3, 100 MHz): δ = 177.90 (s), 151.47 (d, J = 22.6 Hz), 147.12 (d, J = 12.4 Hz), 144.95 (d, J = 21.7 Hz), 141.69 (d, J = 6.5 Hz), 138.86 (d, J = 110.5 Hz), 134.12 (d, J = 86.3 Hz), 133.15 (s), 131.05 (s), 130.88 (s), 130.77 (s), 129.31 (d, J = 13.4 Hz), 128.84 (d, J = 13.8), 128.52 (s), 128.21 (s), 127.43 (s), 118.40 (d, J = 18.0 Hz) ppm; MS (EI): m/z (%): 477.84 (100) [M+], 400.15 (23) [M+ - C6H5], 111.04 (47) [M+ - C14H7S2PO-Br]; HR-MS (TOF MS EI+): calculated for C19H10O2S3PBr m/z: 477.8743 [M+], found m/z: 477.8719, mp: 260 – 263 °C.
4.3 Compound 6 - Yamamoto coupling of 5
0.035 g (0.13 mmol) of Ni(cod)2 was added to a flask containing 0.10 g (0.21 mmol) of the brominated species (5) and 0.034 g (0.13 mmol) of PPh3. 0.014 g (0.134 mmol) of dry cod was added to the flask and was stirred at 60 °C for 72 h. The solution was poured over HCl acidic methanol and filtered. The red precipitate was washed with HCl acidic methanol, ethanol, hot toluene, hot aqueous EDTA (pH = 4), hot aqueous EDTA (pH = 10) and then distilled water (in this order) yielding 6 as a red powder.
Yield: 0.14 g, 0.18 mmol, 82%; 31P-NMR (CDCl3, 162 MHz): δ = 18.85 (s, 1P) ppm; 1H-NMR (CDCl3, 400 MHz): δ = 7.91–7.90 (d, 2H, J = 3.2 Hz), 7.87–7.86 (dd, 2H, J = 3.6 Hz), 7.80–7.75 (m br, 4H), 7.72–7.71 (d, 2H, J = 4.8 Hz), 7.62–7.58 (m br, 2H), 7.52–7.47 (m br, 4H), 7.31–7.30 (d, 2H, J = 2.8 Hz), 7.19–7.17 (m br, 2H,) ppm; MS (ESI): m/z: 794.82 [M+]; HR-MS (MALDI-TOF): calculated for C38H21O4P2S6 m/z: 794.9234 [M + H]+, found m/z: 794.9262, mp: > 300 °C.
CCDC-763125 (3) & 763126 (2x) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request.cif.
Conflict of interest statement
We hereby certify that there is no conflict of interest with this manuscript.
Acknowledgements
Financial support by NSERC of Canada, the Canada School of Energy and Environment, and by the Canada Foundation for Innovation (CFI) is gratefully acknowledged. We thank the University of Calgary for a PURE student scholarship (LDS) and Alberta Ingenuity now part of Alberta Innovates – Technology Futures for New Faculty Awards (CPB and TB).