

1 Introduction
This article concerns compounds consisting of two (or more) monosaccharides linked by formal condensation without using the anomeric centre, creating an ether linkage, as well as their thioether-linked and selenoether-linked counterparts. It would probably be prudent to comment on the nomenclature for such structures, which is not obvious: they have been called disaccharides, diglycoses (anhydrides), pseudodisaccharides, and neodisaccharides, sometimes with the qualifier tail-to-tail. The usage ‘disaccharides’ is incorrect, as this term implies the existence of a glycosidic linkage. ‘Diglycoses’ [1] would appear to be a International Union of Pure and Applied Chemistry (IUPAC) recommended term, but is complicated by the implication that the constituent monosaccharides are not functionalised at the anomeric position. ‘Diglycosides’ or ‘glycosidyl glycoses’ would be the cumbersome extensions of such a nomenclature, although ‘diglycosyl compounds’ may be suitably unspecific regarding the anomeric substituent. ‘Pseudodisaccharides’ is a vague and inclusive term, covering many different sorts of structures judged by its users to be reminiscent of disaccharides. This may include derivatives of carbasugars, pseudocarbasugars, cyclitols, etc., whether glycosidically-linked or not. The structures discussed here can almost certainly be included as pseudodisaccharides. ‘Neodisaccharides’ [2] and ‘Neosaccharides’ [3] are neologisms proposed to describe the specific situation of two monosaccharides linked by condensation without using the anomeric position.
Interest in ether-linked structures has come from two sources: the synthesis of natural products or proposed natural product structures, and the synthesis of proposed glycomimetics. The β-exotoxin I from Bacillus thuringiensis (sometimes called Thuringiensin) contains the unusual d-Glc(5–4)-d-Rib ether linkage (Fig. 1) [4]. The total synthesis of this molecule has been achieved, but the biosynthesis of the ether linkage has yet to be elucidated. A natural product, Coyolosa, extracted from Acrocomia mexicana and found to have hypoglycaemic properties, was assigned an d-All(6–6)-d-All ether-linked structure II and this stimulated a lot of interest from within the synthetic chemistry community. It has since been shown that the proposed structure was not correct, and the actual structure remains unknown [5–7]. Very recently, two further ether-linked neodisaccharides have been reported as substructures of natural products from plants. A d-Glc(3–4)-d-Glc ether III from Palicourea coriacea (a Brazilian flowering shrub, douradinha) [8]; and a d-Glc(2–2)-d-Glc ether IV from Cinnamomum cassia [9]. In both cases, the ether linkage was apparent from three-bond HMBC C–H correlations across the interannular C–O–C bonds. To date, no natural thioether or selenoether neodisaccharide structures have been reported, so interest in these compounds has been entirely from the viewpoint of the synthesis of potential hydrolytically stable disaccharide mimics.
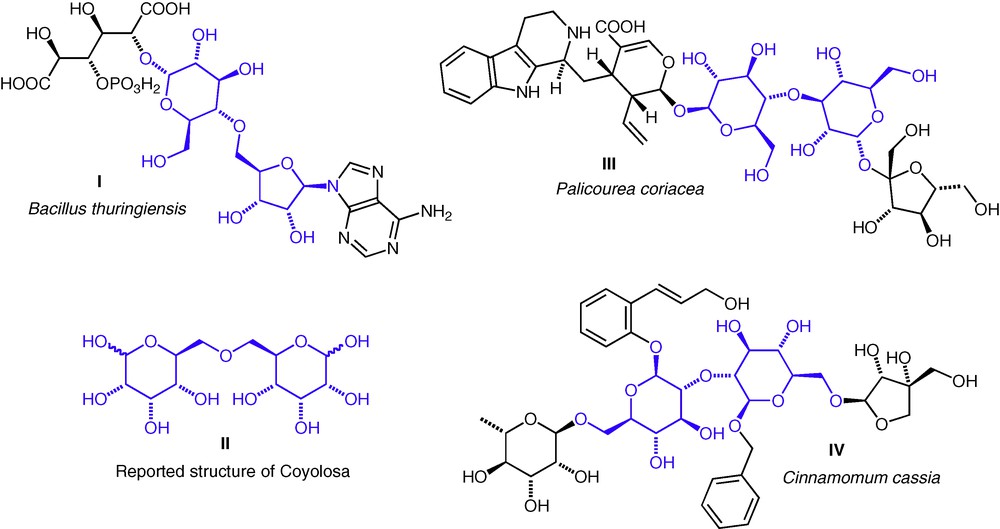
Natural products with neodisaccharide substructures I, III, IV; and the originally proposed structure for Coyolosa II. Neodisaccharide ether substructures are highlighted.
Hydrolytically stable disaccharide mimics are useful tools for glycobiology. The structural similarity between a disaccharide and a neodisaccharide in terms of size, e.g. two pyranose rings linked by a single bridging atom for sec–sec linked structures, and the nature of the functional groups, i.e. polyhydroxylation, has led to speculation that neodisaccharides could mimic disaccharides in their binding to carbohydrate-binding proteins, such as lectins, glycosidases etc. Moreover, the fact that such structures are not acetal-linked would make them less susceptible to hydrolysis by glycosidases or strong acid, so increasing their utility. Exchanging the linking oxygen for other heteroatoms, such as sulfur, selenium or nitrogen, may also alter the properties of the molecule. In the structurally related carbasugar pseudodisaccharide series, it has been shown that a bridging amine results in better inhibition of glycosidases than when the compound is an ether or a thioether [10]. Ether-linked carbadisaccharides have been shown to act as ligands for lectins [11].
However, it was far from obvious that such neodisaccharide structures could act as disaccharide mimics in their binding to carbohydrate-binding proteins. We began our investigation into this area with the following aims:
- • to investigate the synthesis of such structures; ideally, the synthesis would be more straightforward than for similar carbasugar-containing structures that require assembly of a carbasugar monomer;
- • to test the hypothesis that such structures can bind to carbohydrate-binding proteins with similar affinities to disaccharides.
Despite sporadic reports of the synthesis of such structures throughout the last half-century, no report of any interaction with a carbohydrate-binding protein appears to have been published before this year. In this article, I attempt to summarise the state of the art with regard to synthesis and biological properties of neodisaccharides with ether, thioether and selenoether linkages.
2 Ether-linked structures
2.1 Ring-opening of cyclic ethers: epoxides and oxetanes
Epoxides may be opened by nucleophiles under neutral, basic or Lewis acidic conditions. An issue that must be considered is regioselectivity: there are two possible electrophilic carbon atoms, and steric, electronic and stereoelectronic factors may be important in rationalising and predicting which of these carbon atoms is attacked by the nucleophile. Where the electrophilic carbon is secondary, reactions will be stereospecific with inversion of configuration.
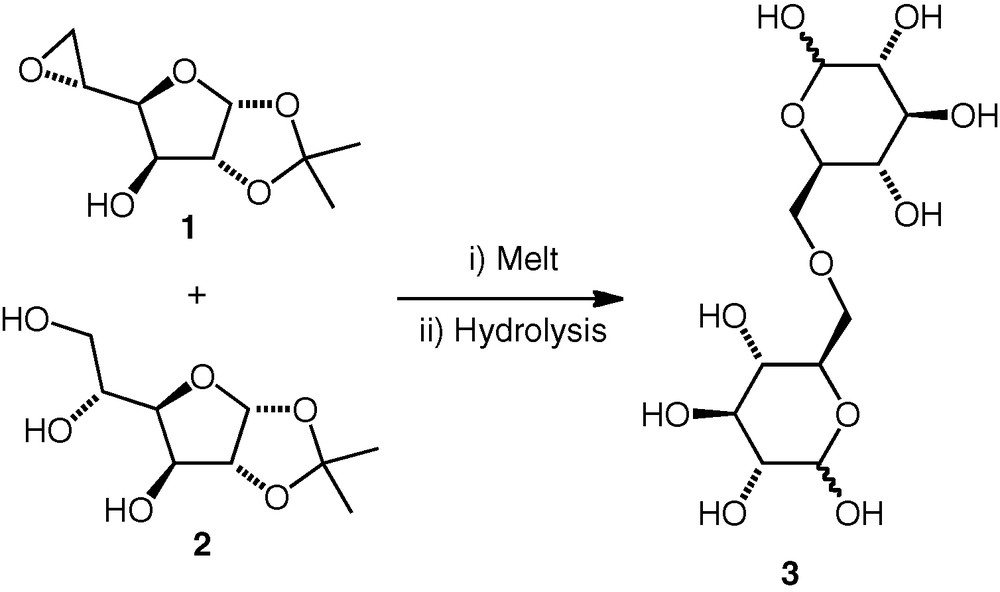
Whistler and Frowein achieved an early example of (6–6)-linked ether formation by heating a (furanose) 5,6-epoxide 1 with a carbohydrate triol 2 (1.4 equiv.) without solvent to 160 °C (Scheme 1) [1]. Under these neutral conditions, only the primary alcohol (OH-6) was reported to react, attacking the epoxide regioselectively at C-6 to give the corresponding ether. When base was added, overalkylation occurred. The isopropylidene protection was removed by acidic hydrolysis. d-Glc(6–6)-d-Glc and d-Glc(6–6)-d-Gal derivatives were synthesised by this method, but Haines reported that his attempts to synthesise a d-All(6–6)-d-All ether by the same method failed [6,7].
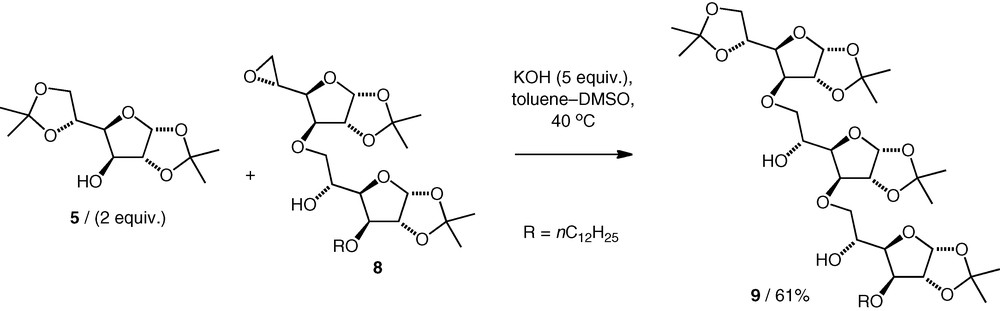
Opening of carbohydrate 5,6-epoxides under basic conditions has been investigated as a method for neodisaccharide formation by Villa, using KOH in DMSO–toluene. A gluco 5,6-epoxide 4 was coupled with primary and secondary carbohydrate alcohols to give (n–6)-linked ethers (e.g. 6) (Scheme 2) [12,13]. When equimolar amounts of the alcohol and epoxide were used, higher homologues were observed, viz triglycosyl (e.g. 7) and tetraglycosyl compounds (di:tri:tetra, 60:33:7) arising from the nucleophilic attack of the neodisaccharide product alcohol on another molecule of the epoxide. When excess alcohol starting material was used, the quantities of higher homologues decreased. Hence, when 2.5 equiv. of the alcohol nucleophiles were used, diglycosyl ethers were formed in 61–79% yield, with no apparent difference between the performance of the reactions of primary versus secondary carbohydrate alcohols.
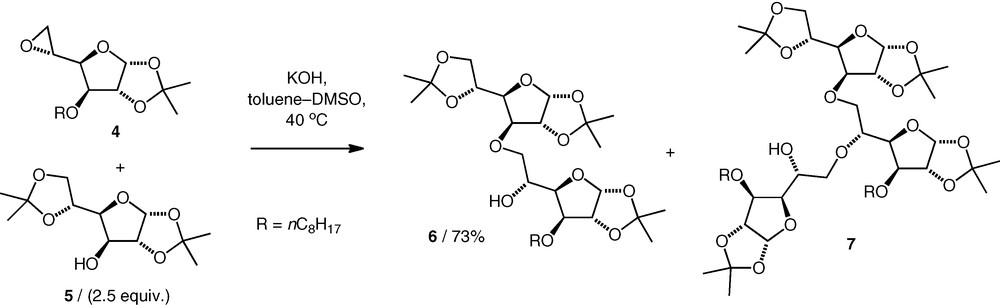
Other 5,6-epoxides were also found to react with alcohols under the same basic conditions. A neotrisaccharide 9 was obtained by coupling of a neodisaccharide 5,6-epoxide 8 with an excess of diacetone d-glucose 5 (2 equiv.) in 61% yield (Scheme 3) [14], while coupling of a d-mannose 5,6-epoxide with a d-xylose OH-3 alcohol (2 equiv.) gave a d-Xyl(3–6)-d-Man neodisaccharide (74%) along with a small amount (8%) of the neotrisaccharide [15].
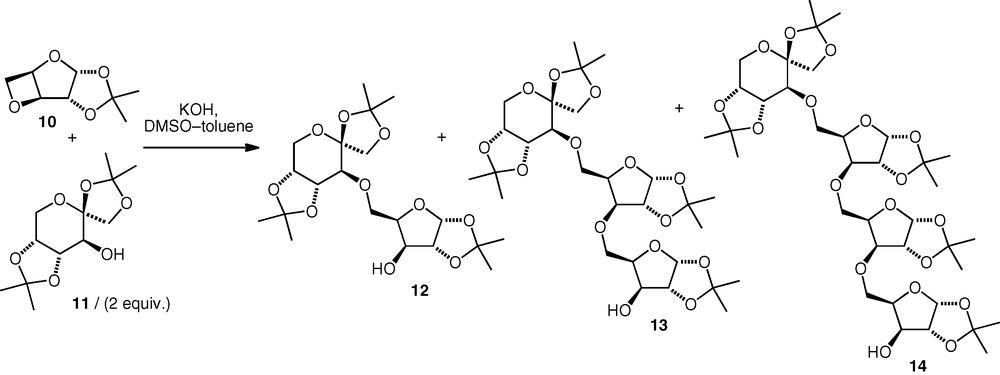
In a related approach, 3,5-anhydro-d-xylose derivatives (i.e. oxetanes) have also been used as electrophiles under the same basic conditions [15,16]. The oxetane 10 reacted with alcohols with exclusive regioselective attack at the sterically more accessible primary carbon (C-5). Depending on the ratio of alcohol to electrophile, di- (12) tri- (13) or tetraglycosyl (14) derivatives were formed (Scheme 4). The proportions of the products were found to be independent of solvent composition and concentration, but depend only on the ratio of reactants. The optimum yield of neodisaccharide was obtained (e.g. 44% isolated diglycosyl; di:tri:tetra, 77:20:3) when excess alcohol was used, while the use of equimolar quantities of nucleophile and electrophile resulted in the formation of more of the higher oligomers. Essentially, no difference was observed between the performances of the reactions of primary versus secondary carbohydrate versus noncarbohydrate alcohols.
All the ring-opening reactions discussed so far proceeded with regioselective nucleophilic attack at a primary carbon, presumably on steric grounds. When an epoxide electrophile in which both of the epoxide carbon atoms are secondary is used, the problem of regioselectivity becomes more significant. In six-membered rings such as those described below, stereoelectronic effects leading to trans-diaxial ring-opened products tend to dominate, especially where the six-membered ring is rendered inflexible by the presence of a second ring, as is the case in 1,6-anhydrosugars or 4,6-benzylidene-protected sugars.
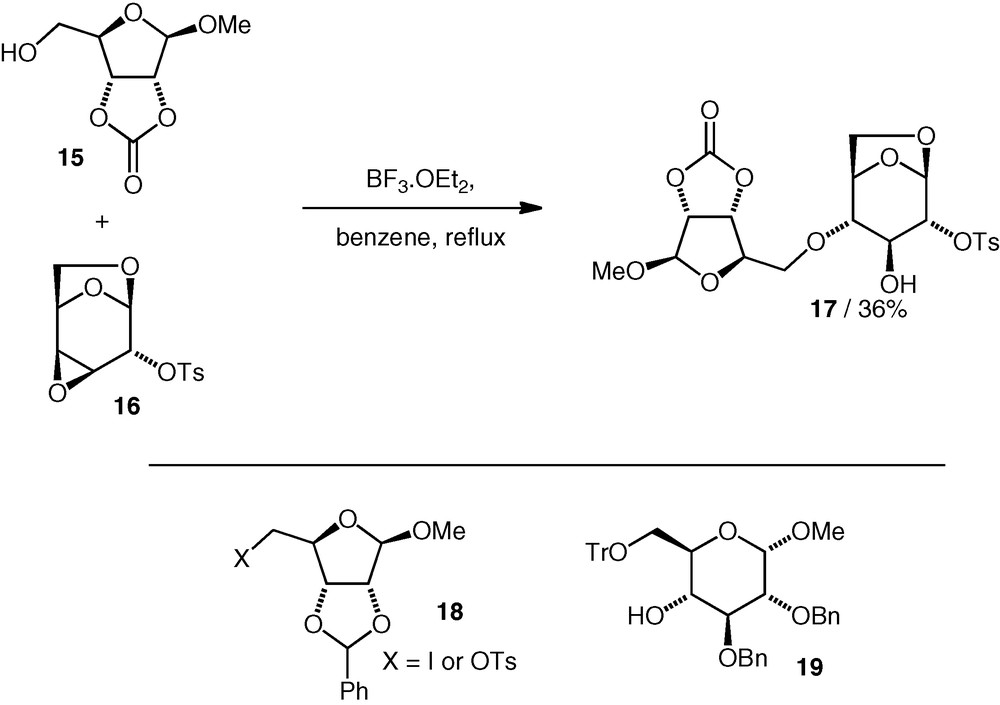
The successful assembly of the neodisaccharide from B. thuringiensis, i.e. d-Rib(5–4)-d-Glc, was accomplished by Prystas and Sorm using an epoxide-opening approach under Lewis acidic conditions [17]. A primary d-ribose (OH-5) nucleophile 15 attacked a conformationally locked d-galacto 3,4-epoxide 16 regioselectively at C-4 to give the trans-diaxial opening product 17 (Scheme 5). Variation of the protecting groups on the nucleophile affected the reaction yield. An attempt to form the ether bond by alkylation of the secondary OH-4 d-gluco alcohol 19 using C-6 iodide or C-6 tosylate 18 failed to give any coupling product (cf. alkylation, vide infra). Some further variants of the coupling reaction to achieve the d-Rib(5–4)-d-Glc linkage based on Lewis acid catalysed opening of 1,6-anhydrosugar 3,4-epoxides have also been published [18,19].
Variously protected carbohydrate 2,3-epoxides have been investigated in coupling reactions with a primary (OH-6) carbohydrate alcohol 21 under both basic and Lewis acidic conditions (Scheme 6) [20]. The α-d-manno epoxides 20 were opened at C-3 exclusively under basic conditions (10 equiv. of alcohol was used) to give α-d-altroside ethers (22) in up to 68–89% yield, and no difference between the outcome of the reactions of the benzyl-protected and benzylidene-protected epoxides was observed. It should be mentioned that the pyranose ring of such α-d-altrosides may be conformationally flexible when it is not locked by a cyclic 4,6-protecting group [21] so O-2 and O-3 should possibly not be described as having a trans-diaxial arrangement in such circumstances. Having said that, the trans-diaxial conformation should be energetically more accessible for an α-d-altroside than for an α-d-glucoside. Under Lewis acidic conditions, the regioselectivity was unchanged: 3-substituted α-d-altrosides 22 were formed exclusively with the OH-6 alcohol 21 (10 equiv.). A secondary alcohol (glucose OH-4) failed to open the benzyl-ether-protected d-manno epoxide 20 under Lewis acidic conditions.
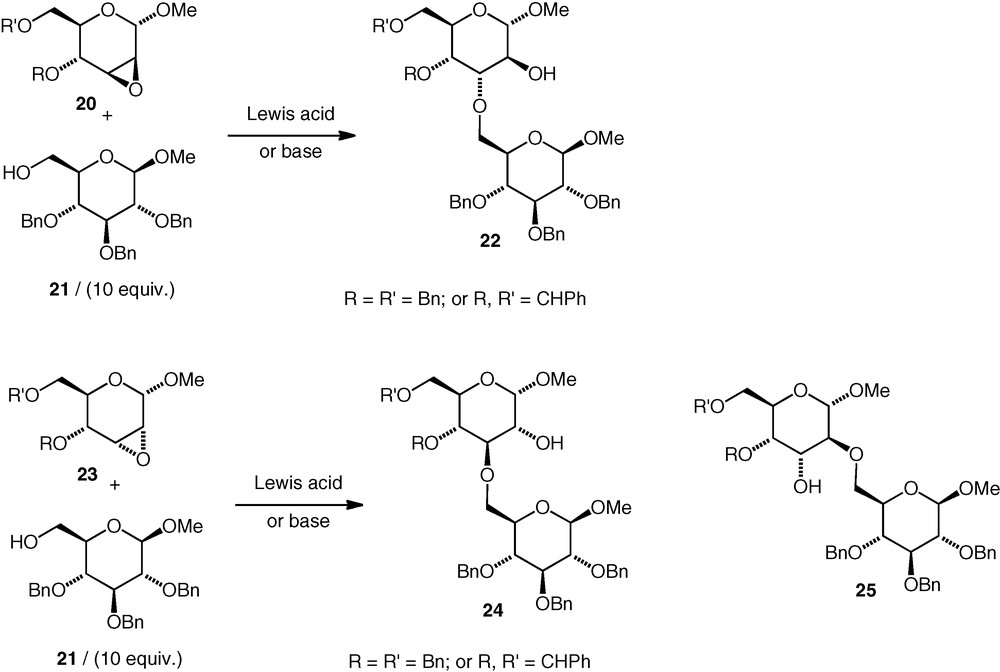
The α-d-allo epoxides 23 were opened under basic conditions (with 10 equiv. alcohol) with rather worse regioselectivity. Both (3–6)-linked 24 (α-d-gluco) and (2–6)-linked 25 (α-d-altro) ethers were formed (59–95% total yield). The regioselectivity was better (d-altro:d-gluco ca 8:1) when a cyclic 4,6-protecting group rigidified the pyranoside ring than when benzyl ethers were used (d-altro:d-gluco ca 1:1). Under Lewis acidic conditions, reactions with d-allo epoxides 23 gave multiple products, and only low yields of the neodisaccharide ethers were seen. Deprotection of the (2–6) and (3–6)-linked ethers made by this approach was not reported.
2.2 Reductive etherification
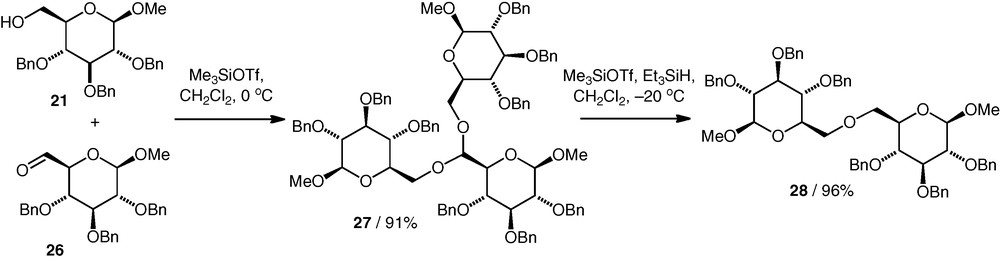
A reductive etherification approach to (6–6)-linked neodisaccharide ethers was developed by Takahashi et al. [5] to try to identify the natural product Coyolosa. In this method, a C-6 aldehyde 26 condensed with two molecules of OH-6 alcohol 21 to give an acetal 27 (Scheme 7). The acetal then underwent Lewis acid catalysed hydride reduction to give the ether 28. A number of C2-symmetrical and unsymmetrical ethers based on different monosaccharide components were synthesised in this way. Only two equivalents of the alcohol are formally required for acetal formation, one of which is then released during the reduction step. In practice though, 10 equiv. alcohol were needed to obtain optimised acetalisation yields of 69–93%. The reduction step gave the ethers in yields of 56–96%. A one-pot approach was also developed, which gave the ether directly from the aldehyde and alcohol in up to 92% yield. All the ether diglycoses were prepared perbenzylated. The benzyl ether protection was removed by catalytic hydrogenolysis, which was followed by acidic hydrolysis of the methyl glycoside moieties to give the doubly reducing Coyolosa candidates.
The reductive etherification method has also been applied to the synthesis of (2–6)-linked and (3–6)-linked neodisaccharides, based on the reaction of a C-6 aldehyde with either an OH-2 alcohol (excess) followed by reduction, or a 2,3-diol followed by regioselective reduction of the cyclic acetal, respectively [3].
2.3 Alkylation
Probably, the most conceptually simple etherification reactions are Williamson type reactions, i.e. alkylation of alcohols under basic conditions. SN2 reactions are less favourable on carbohydrates than on simple alkyl halides, and it may be expected that competing elimination reactions would hinder SN2 processes from being an efficient route to ether neodisaccharides, given the strongly basic nature of the alkoxide nucleophiles. However, some success using this approach has been achieved.
Haines has coupled C-6 triflates (e.g. 30) with OH-6 alcohols (e.g. 29) to construct ether linkages (e.g. 31) under basic conditions (Scheme 8) [6,7]. C2-symmetrical (6–6)-linked di-d-allose, di-d-galactose, di-d-glucose and di-d-mannose ethers were obtained in 79–98% yields using a slight excess of the triflate (1–1.3 equiv.). The protecting groups were removed to give the unprotected compounds, none of which appear to be the elusive Coyolosa. Where the neodisaccharide was synthesised as its methyl glycoside(s), the anomeric centre(s) could be deprotected by acidic hydrolysis [7]. It is noteworthy that this approach with C-6 triflates was successful whereas related attempts with C-6 tosylates or C-6 iodides failed to perform as well, [5] which is in agreement with the earlier results of Sorm and Prystas, vide supra. A C-6 triflate was also used to alkylate OH-2 of a d-gluconic acid derivative, the ether being an intermediate in the synthesis of truncated neuraminic acid glycosides [22].
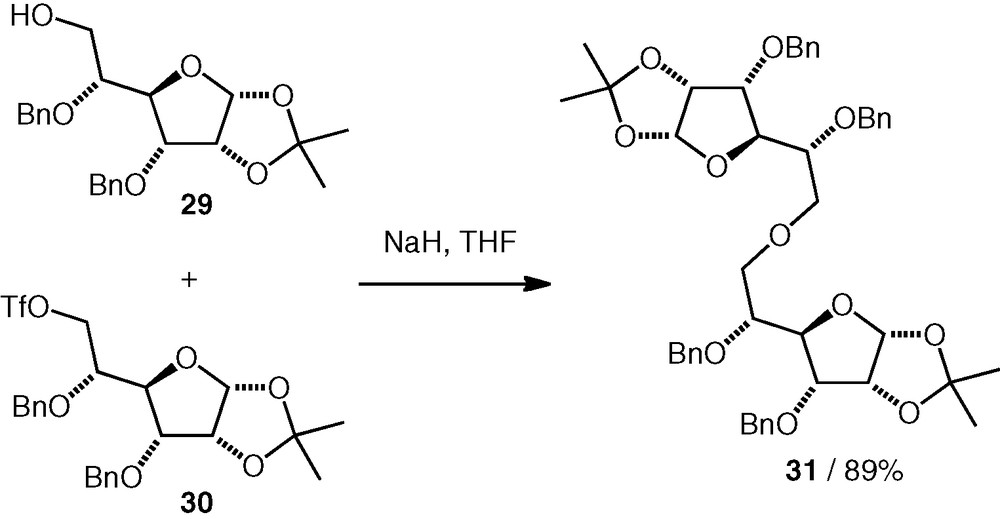
In our group, we have studied alkylation reactions using a secondary d-allo C-3 triflate 32 [23]. This compound underwent coupling reactions with various secondary carbohydrate alcohols (1.5–3.3 equiv. of triflate was used) to give (n–3)-linked ethers (e.g. 34, Scheme 9), often in good yields 63–95%. We sometimes isolated d-Glc(3–3)-d-Glc and d-All(3–3)-d-Glc ether by-products formed by dimerisation of the triflate 32.
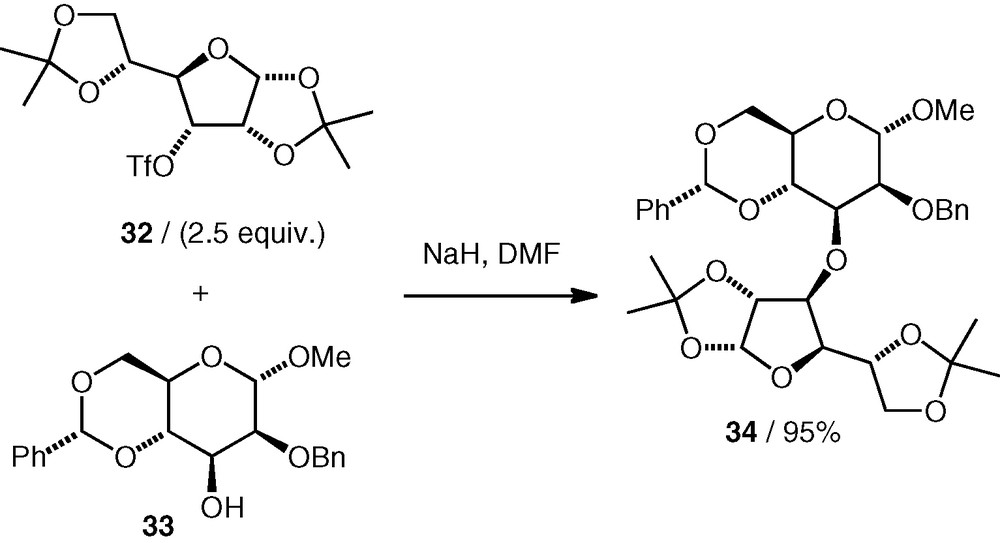
Considering the positive results that have been found with the C-6 and C-3 triflates described here, it seems likely that other triflates exist or can be made for which competing elimination would also not be a problem. The C-3 d-allo triflate 32 and C-6 d-gluco triflate 35 have both been used before as alkylating agents for oxygen nucleophiles in the synthesis of glycosides (from hemiacetal nucleophiles) [24] or pseudodisaccharides (from cyclitol or carbasugar nucleophiles) [25]; The C-3 d-gulo triflate 36 [24] and C-4 d-galacto triflate 37 [24,25] (Fig. 2) have been used in similar reactions, so these are examples of compounds that may offer routes to 3-substituted d-galactose and 4-substituted d-glucose pseudodisaccharide ethers, respectively.
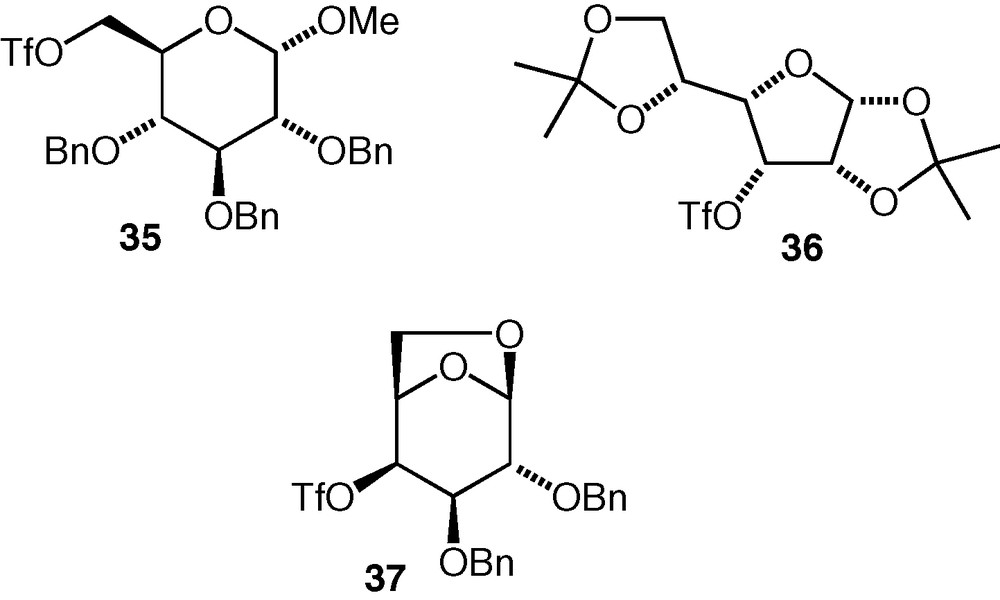
Some carbohydrate triflates with potential for use as electrophiles in etherification reactions.
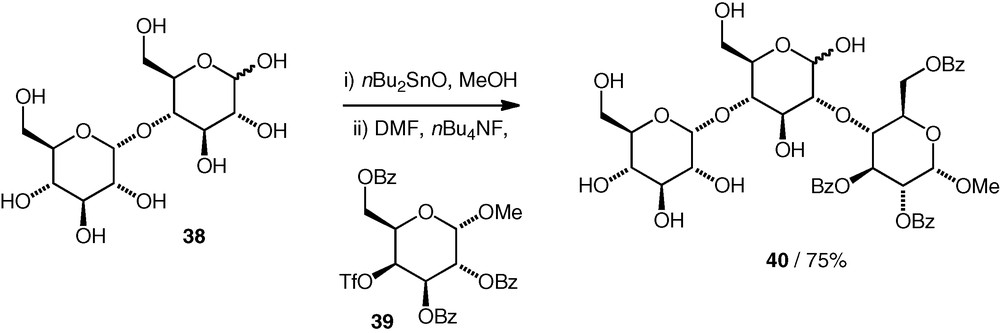
The formation of ether-linked pseudo-di- or -trisaccharides has been observed in coupling reactions between tin acetals (as nucleophiles) and carbohydrate triflates [26]. When maltose 38 was used as a nucleophile, coupling with a galacto C-4 triflate 39 occurred with inversion of configuration at C-4 of the electrophile to give a pseudotrisaccharide 40 containing a d-Glc(2–4)-d-Glc substructure as the major product (75%, Scheme 10). Presumably, the equatorial OH-2 in a glucose 1,2-stannylene is more nucleophilic than the axial OH-1. Some other examples were seen, including a d-Man(3–6)-d-Glc structure, but usually, the (desired) attack of O-1 on the electrophile dominated, resulting in glycosidic products.
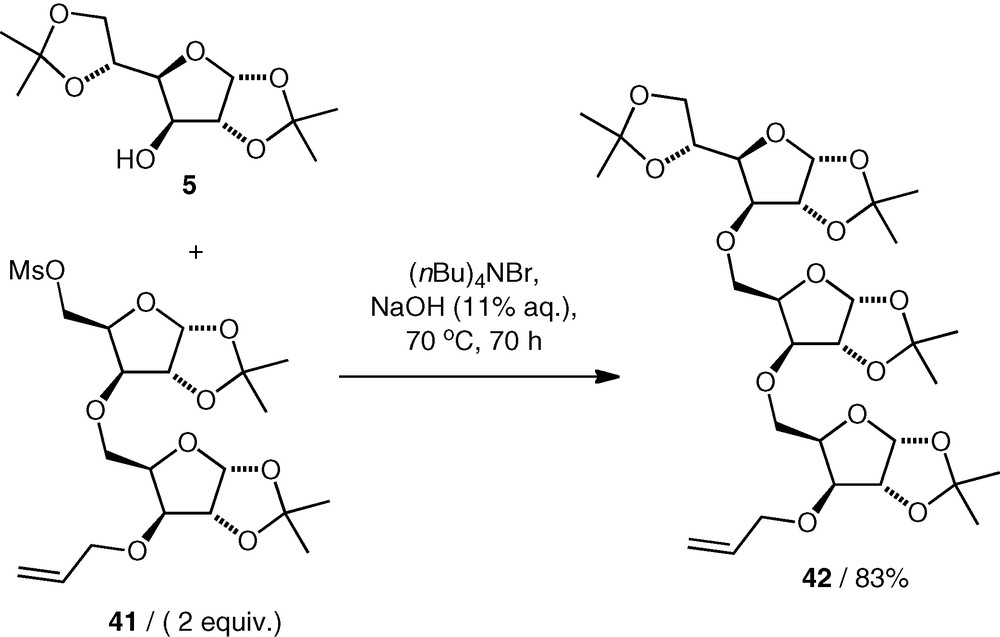
Reaction conditions have been found that enable the use of primary mesylates as alkylating agents (rather than expensive and reactive triflates) in etherification reactions. C-5 mesylates of pentose derivatives, e.g. 41, were coupled with nucleophiles under basic conditions in water (NaOH 11%, (nBu)4NBr with heating for extended time) to give the ethers (e.g. 42) in good yield (Scheme 11) [27–29]. An excess of the mesylate was required, and the elimination product was also observed. Primary and secondary carbohydrate alcohols were both used as nucleophiles, giving (5–5)-linked [27] and (3–5)-linked [28,29] ethers.
3 Thioether-linked structures
3.1 Ring-opening of epoxides
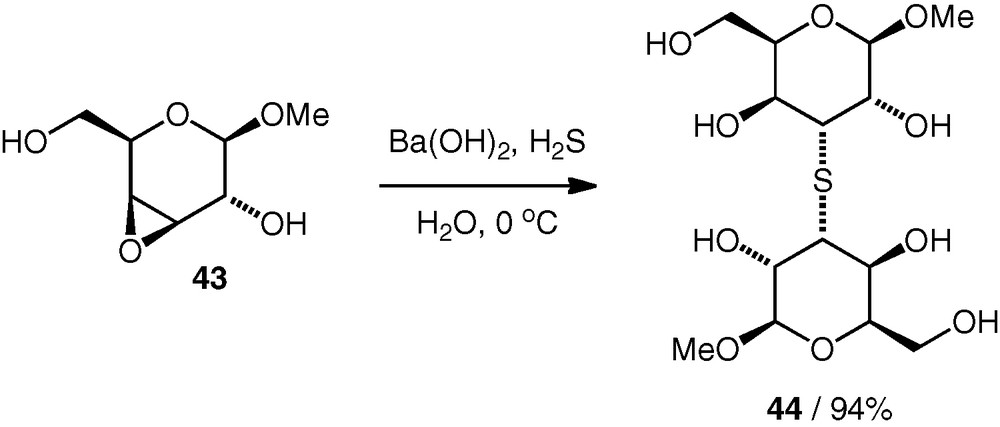
Epoxide-opening reactions leading to thioethers have also been reported. In the 1960 s, Dahlgard reported the dimerisation of a d-galacto 3,4-epoxide 43 using a divalent sulfur nucleophile (from Ba(OH)2 and H2S, i.e. under basic conditions) with regioselective attack at C-3 to give an unprotected (S3–3)-linked thioether pseudodisaccharide 44 in excellent yield (Scheme 12) [30].
A single example of neodisaccharide formation by epoxide-opening with a nucleophilic thiol under Lewis acidic conditions has been demonstrated [20]. An α-d-manno 2,3-epoxide 20 (R = R′ = Bn) was attacked regioselectively at C-3 by a primary (C-6) thiol, giving the (S3–6)-linked α-d-altroside thioether as sole product.
3.2 Michael addition
Conjugate additions of carbohydrate thiols to α,β-unsaturated ketones or α,β-unsaturated lactones have been used to construct thioether linkages between monosaccharides. An issue that must be considered in such reactions is diastereoselectivity. The thioether products resulting from such couplings retain the carbonyl group in an oxidised state; also, one of the monosaccharide components of the thioether products will be a deoxysugar, lacking functionality at the position between the carbonyl group and the new C–S bond.
Sridhar et al. have described the synthesis of a single (S3–6)-linked derivative by attack of a C-6 thiol (formed in situ from the disulfide) on a 2,3-unsaturated lactone [31]. The reaction used equimolar amounts of the two carbohydrate components, and was promoted by an ammonium MoS42− salt. The coupled product was isolated as a single diastereomer with an axial C–S bond in 90% yield. This compound was not deprotected.
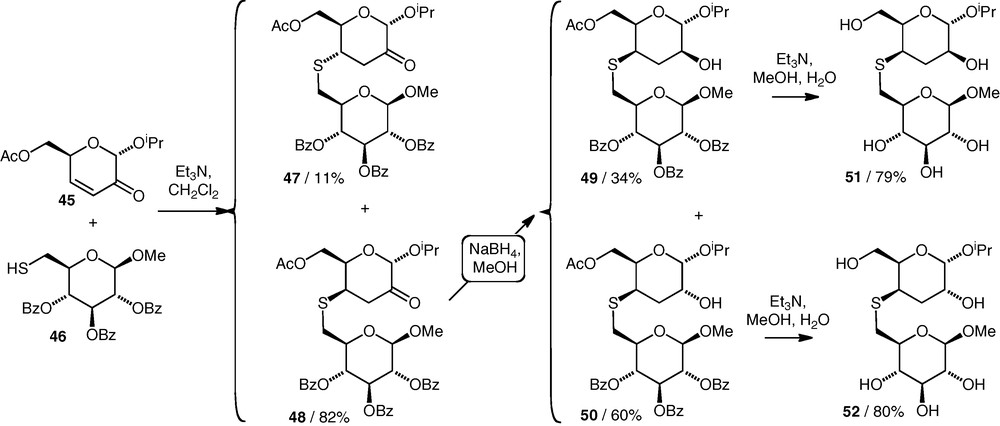
Uhrig et al. have described the coupling reactions of two primary (C-6) thiol nucleophiles (e.g. 46) with the 3,4-unsaturated 2-ketosugar 45 (Scheme 13) [32]. Both gave (S4–6)-linked thioethers (e.g. 47 and 48) in high yields from equimolar amounts of the components, with good diastereoselectivity in favour of the product with an axial C–S bond (48). The ketones were then reduced and all compounds were deprotected to give the free thioethers 51 and 52. It was stated that the thioethers would be tested as glycosidase inhibitors, but no data have been published to date.
3.3 Alkylation
Alkylation by displacement of sulfonates under SN2 reaction conditions has the advantage of offering a stereospecific reaction (with inversion of configuration) and also regiospecific reaction. A disadvantage is that competing elimination processes or other side reactions can sometimes decrease the yield of coupling product.
SN2 displacement of carbohydrate triflates by thiol nucleophiles has been fairly widely investigated in the context of the synthesis of thiooligosaccharides [33]. In our group, we have looked at the synthesis of neodisaccharides by two strategies. Firstly, a stepwise approach based on (i) introduction of sulfur into a carbohydrate by displacement of a sulfonate leaving group with thioacetate; (ii) removal of the acetate to give the free carbohydrate thiol; (iii) coupling of the carbohydrate thiol with a second carbohydrate sulfonate to give the neodisaccharide thioether (Scheme 14a). This approach was used for the synthesis of both C2-symmetrical and unsymmetrical thioethers [34,35]. Using a slight excess (typically 1.5 equiv.) of either the thiol or the triflate, thioethers were formed in 42–94% yield. Alternatively, carbohydrate triflates were coupled with a divalent sulfur nucleophile (from Na2S) to give C2-symmetrical neodisaccharides in a single step, often in very good yields (Scheme 14b) [35].
The sulfonates we used are shown in Fig. 3. Triflates were used at secondary carbons (i.e. 32, 54–61), and mesylates at primary carbons (i.e. 53). For some of the triflates investigated (32 and 54–56), the substitution reactions both with potassium thioacetate and with carbohydrate thiolate nucleophiles occurred cleanly with inversion of configuration and with no by-products resulting from competing elimination reactions. For some others (57–59), significant elimination was observed in substitution reactions with thioacetate, and the yields of substitution products were lower. Finally, some triflates (60, 61) failed to give useful amounts of the substitution products in reactions with potassium thioacetate due to extensive decomposition of the triflate under the reaction conditions. For those cases, where elimination was not a problem, sec–sec linked thioethers could be formed as straightforwardly as primary–sec and primary–primary thioethers.
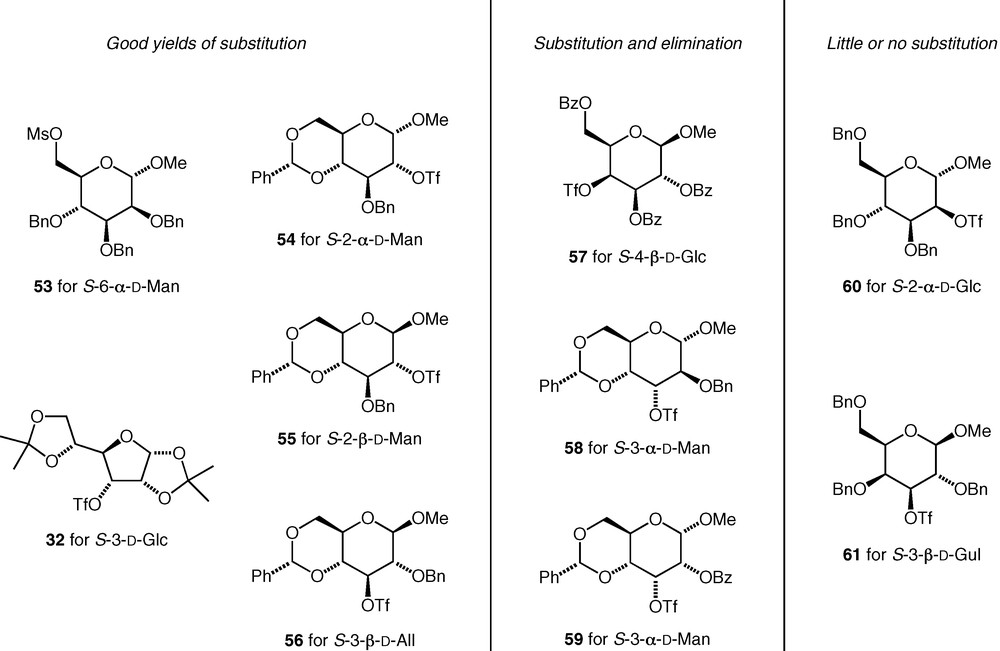
Carbohydrate sulfonates tested in C–S bond forming reactions.
All of the thioethers synthesised could be deprotected using Birch conditions (Na, NH3 (l)) to remove benzyl ethers and benzylidene acetals, acidic hydrolysis to remove isopropylidene acetals, and methanolysis (NaOMe) to remove esters [35].
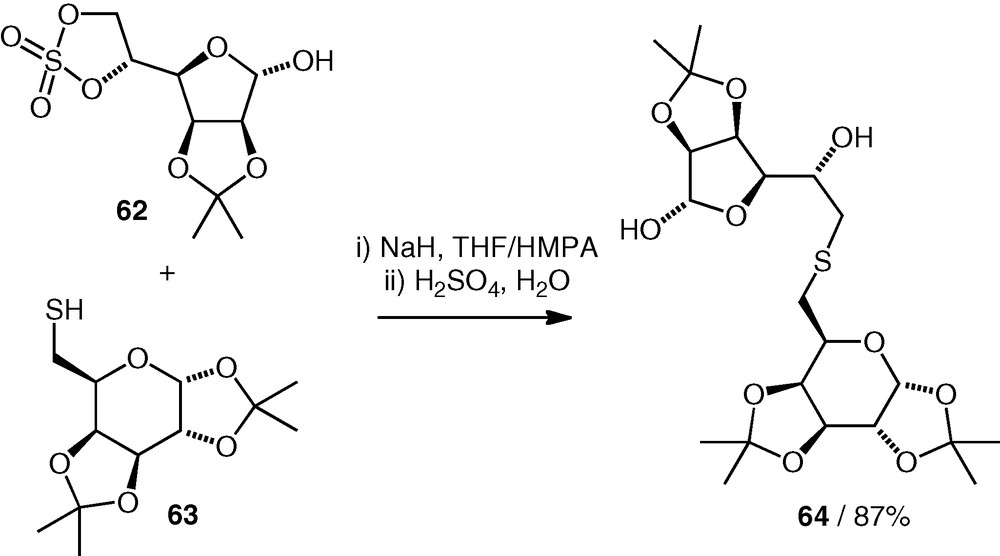
A carbohydrate sulfate has been used by Wadouachi et al. as an electrophile for thioether neodisaccharide formation [36]. An isopropylidene-protected d-Gal(S6–6)-d-Man structure 64 was obtained by reaction of a thiol 63 (2 equiv.) with a cyclic 5,6-sulfate 62 under basic conditions (Scheme 15). The reaction occurred regioselectively at the primary carbon of the electrophile, and the neodisaccharide product was not deprotected.
4 Selenoether-linked structures
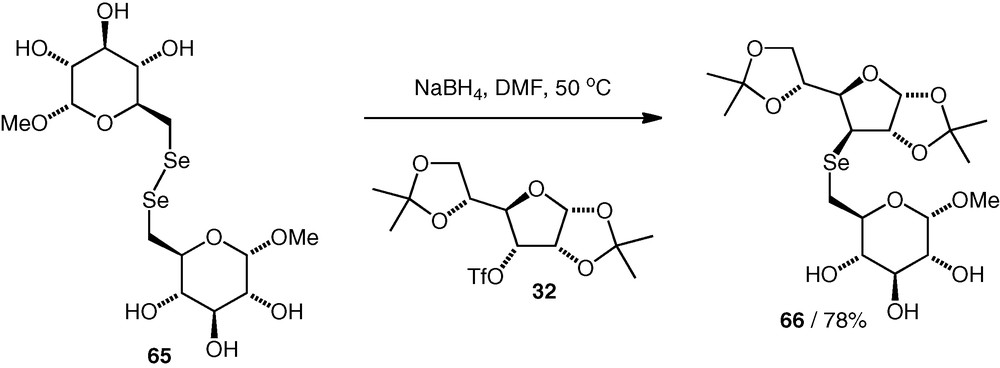
In our group, we have also looked at selenoethers as potential disaccharide analogues [37], and synthesised (Se3–6)- and (Se3–3)-linked compounds. Selenols are easily oxidised to diselenides, and we found that the optimum procedure involved generation of a selenolate nucleophile in situ by reduction of a carbohydrate diselenide (e.g. 65) in the presence of a carbohydrate triflate 32 (1.2–1.3 equiv.). In this way, triflate displacement could occur without isolation of the selenol, and the selenoethers (e.g. 66) were formed in 78–87% yield with clean inversion of configuration at the site of substitution. The presence of selenoethers in the molecules was incompatible with deprotection of benzyl ethers under Birch conditions, so instead, we synthesised the selenoether neodisaccharide with minimal protecting groups in place (Scheme 16). Isopropylidene protection was easily removed by acidic hydrolysis.
5 Biological activity
5.1 Hypoglycaemic properties
Because Coyolosa was reported to have hypoglycaemic properties, the synthetic candidates synthesised by Takahashi et al., all (6–6)-linked ethers, were assayed for this property in rats in vivo [5]. The d-Man(6–6)-d-Man derivative was found to behave similarly to Coyolosa (although the physical data for the compound and its acetate reveal that the compounds are not identical).
5.2 Binding to lectins
A number (25) of our synthetic thioether, ether and selenoether-linked compounds were tested as potential ligands for the plant lectins banana lectin and Concanavalin A [35]. The results may be briefly summarised as follows.
The binding affinities of the neodisaccharides for each of the two lectins varied widely, with many compounds not binding at all within the limit of detection (using ITC microcalorimetry). We found neodisaccharide ligands for the banana lectin that bound with affinities higher than for monosaccharides, but even the best binding compounds did not have as high affinity for the lectin as strongly binding disaccharides (e.g. d-Glc(β1 → 3)-d-GlcαMe, Ka 2400 M−1). For Concanavalin A, we found neodisaccharides that bound to the lectin with affinities similar to and in excess of that of the strongest known binding natural disaccharide ligand (i.e. d-Man(α1 → 2)-d-Man, Ka 35,700 M−1). Some of the best ligands for the two lectins are shown in Fig. 4.
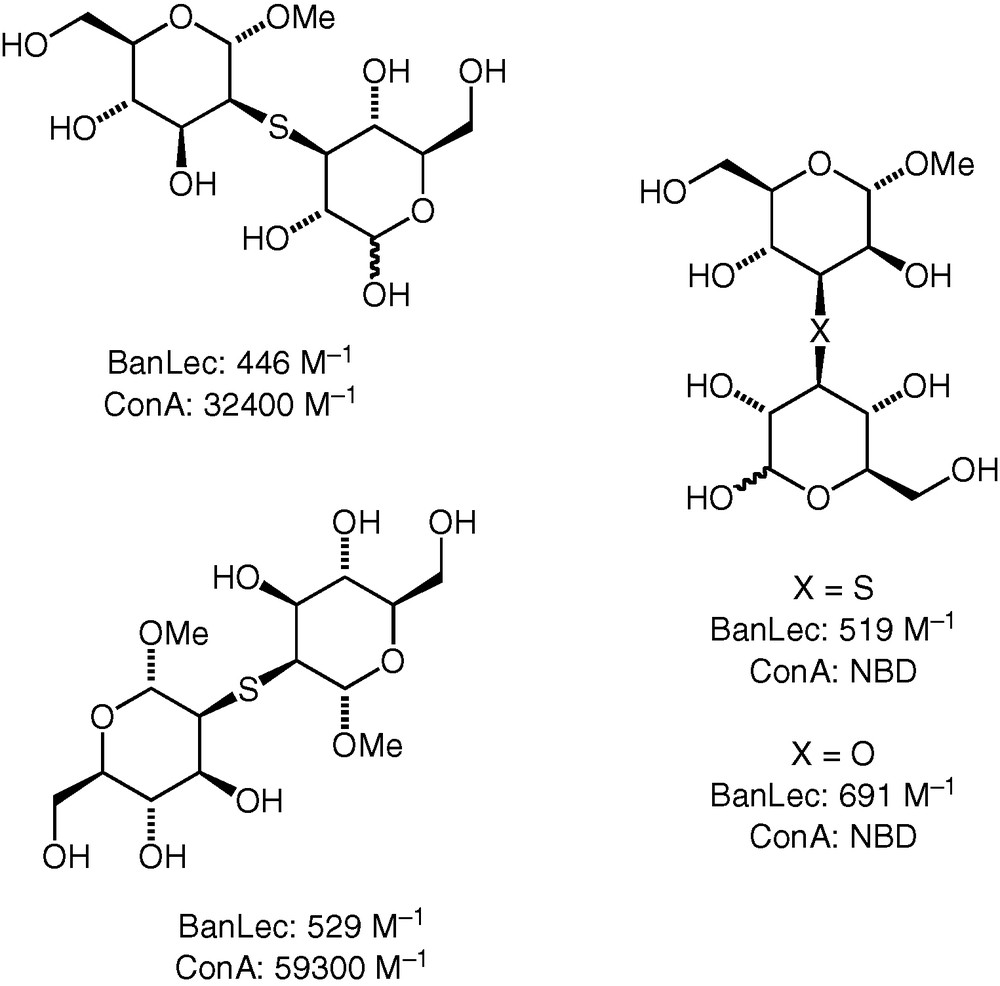
Some diglycosyl ethers and thioethers with higher lectin binding affinity. NBD : no binding detected.
Ethers and thioethers both bound to the lectins, and in some cases, it was possible to make a direct comparison between two neodisaccharides differing only in the bridging atom. Apparently, the identity of the bridging atom does affect the binding affinity, albeit to a lesser extent than the identity of the monosaccharide constituents. Oxidation of the thioether bridging sulfur to sulfoxide and sulfone levels was also investigated for some of the compounds and was found to have an effect on the affinity. A more detailed analysis of the binding event and of the possible mimicry of the binding of a natural substrate remains to be done.
6 Future prospects
One possible future direction for research would be to search for ligands for other lectins or carbohydrate-binding proteins of biological relevance, such as pathogenic adhesins or galectins. Structural studies to rationalise the lectin–ligand interactions would give valuable information, as would an examination of the conformational behaviour of the ether or thioether interannular bonds. It is possible that the higher than usual number (i.e. higher than in a glycoside) of hydroxyl groups flanking the linking bond could lead to more interresidue hydrogen bonding. In terms of synthesis, the majority of the neodisaccharide compounds that have been synthesised involve at least one primary carbon centre in the linkage, i.e. C-6 in aldohexoses and C-5 in pentoses. Further work on the synthesis of compounds with linkages at secondary carbons would be welcome.
Acknowledgement
I am grateful to Viviane Fournière of the University of Bangor in Wales, UK, for her help with the French language.