

1 Introduction
Heparan sulfate (HS) and heparin (HP), like other glycosaminoglycan such as hyaluronate, chondroitin sulfate, dermatan sulfate and keratan sulfate, are linear and sulfated polysaccharides in which a basic disaccharide is repeated along the chain. HS and HP share a common backbone containing d-glucosaminyl moieties and thus fall in the glucosaminoglycan category. Structural distinction between HS and HP is difficult to establish and is generally performed on the basis of the polysaccharide sulfate content, since in HP a high proportion, generally over 80%, of the glucosaminyl residues is N-sulfated [1]. The major difference between both polymers is, in fact, not structural and is better established based on their localisation in vertebrates: HP is essentially confined to connective tissue type mast-cells and is released in the extracellular matrix free from any core protein. In contrast, HS is present linked to a core protein at the cell surface or in the extracellular matrix surrounding virtually any type of cell [1].
2 Sulfation and epimerisation: a first molecular diversity level
The basic disaccharide unit of HS/HP consists in an uronic acid linked, in a 1,2-trans stereochemistry, to the position 4 of the α-d-glycosaminyl moiety linked itself to the position 4 of the next uronic acid residue (Fig. 1). Although such a linear repetitive backbone should lead to a limited molecular diversity, HS/HP is one of the most heterogeneous biopolymers: the uronic acid may have either the d-gluco or l-ido configuration while various sulfation patterns (sulfoforms) may occur along the chain [2–4]. O-sulfation may occur on position 2 of the uronic acid and 3 and/or 6 of the amino-sugar, while the glucosamine nitrogen may be sulfated, acetylated or, less frequently, unmodified, leading to 48 possible disaccharides (Fig. 1). Based on current knowledge on HS biosynthesis, all the theoretical disaccharide structures cannot be expressed, however, 23 of them have already been characterised [5]. The diversity grows exponentially with the polymer length, leading to 2304 possible tetrasaccharides, 110,592 hexasaccharides and more than 5 × 106 octasaccharides (Fig. 1). In HS, hypervariable domains of great complexity are thus formed along the polymer, providing numerous docking sites for protein ligands, enabling selective interaction in a topologically and temporally controlled manner. Analysis of HS from different mammalian tissues revealed the tissue-specific composition of samples, pointing to strict regulation of biosynthetic polymer modification with the presumed goal of generating heterogeneous sequences with biological specificity [6–7]. It has been indeed shown in mice that cell surface HS composition profile was dependent of the tissue from which it was extracted but was superimposable for tissue corresponding samples from different individuals [8]. With respect to structural diversity HP, which is the result of extensive biosynthetic polymer modification, may be conceived as an over-sulfated HS. HP chains are less heterogeneous than HS ones and contain large homogenous domains of repeating IdoUA-2-O-SO3Na-GlcNHSO3Na-6-O-SO3Na, referred as the regular repetitive region of HP [5].
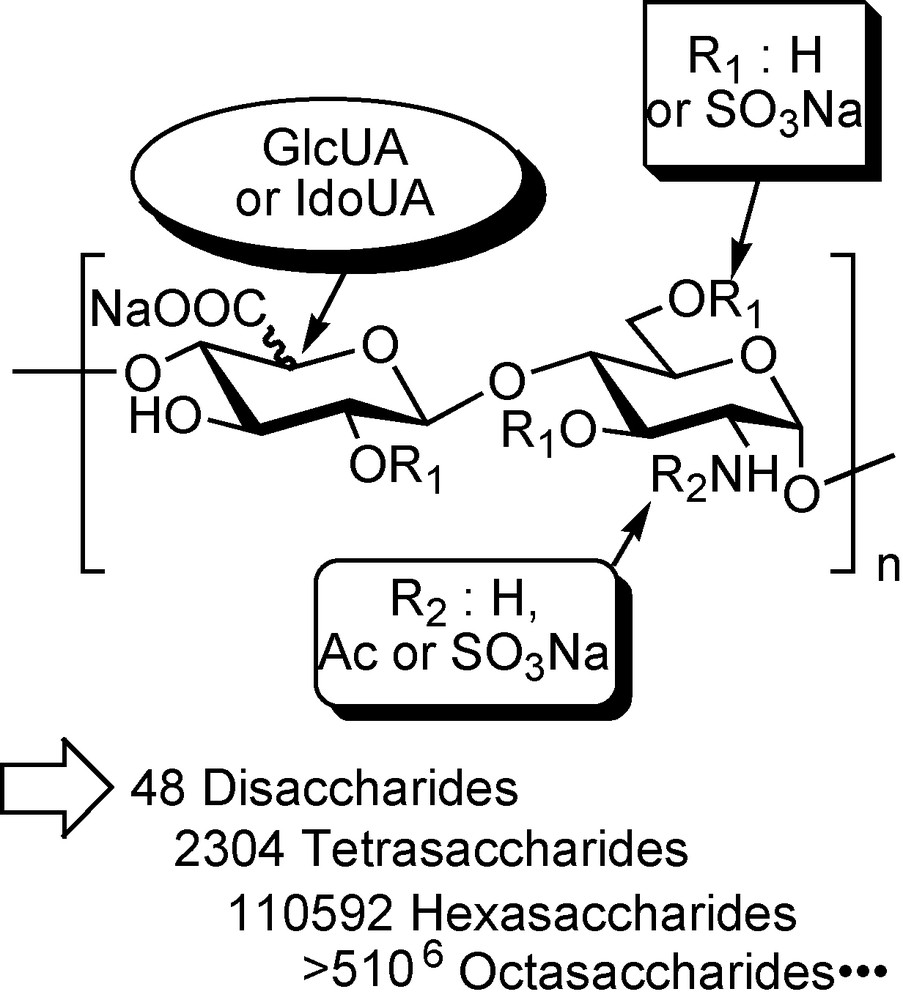
Theoretical molecular diversity found in HS/HP.
At the cell surface or in the extracellular matrix, HS chains interact and regulate the activity of numerous extracellular proteins such as growth factors, cytokines, chemokines, viral proteins or coagulation factors [2,9–10]. Although HP is not exposed at the cell surface, it also binds HS binding protein, often with highest affinity due to its higher charge density, however, often with less specificity due to its higher structural homogeneity. It may thus seems contradictory that, to date, the sole fully characterised GAG-protein interaction is the HP/antithrombin-III (AT-III) one, which is at the origin of the well-known anti-thrombotic properties of HP. In this case, the anti-Xa property of heparin was attributed to the presence in the chain of a specific structure: a pentasaccharide of relatively high occurrence in HP, with precise uronic and sulfation patterns, that confers a high affinity and specificity to AT III and promotes the conformational change catalysing the inhibition of numerous coagulation factors by AT-III [11]. The chemical synthesis of this pentasaccharide has led to the development of Arixtra® 1, a FDA and EMEA-approved drug against deep vein thrombosis (Fig. 2) [12].
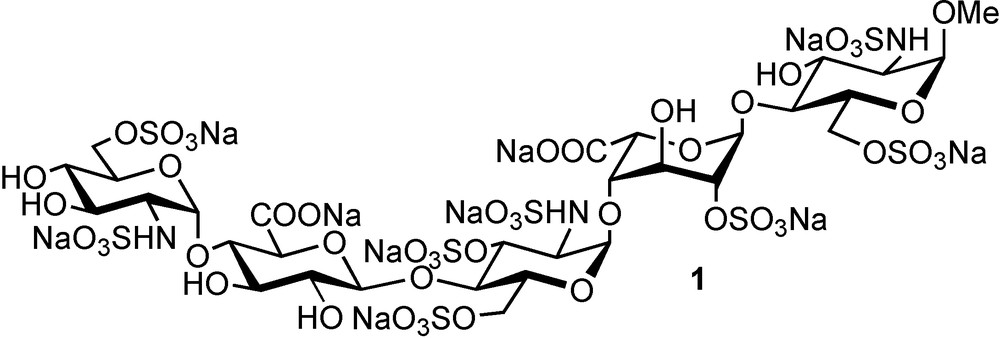
Structure of the active principle of Arixtra® (fondaparinux sodium salt).
From the first heroic synthesis in the mid 1980s [13], impressive and innovative synthetic efforts have been performed in order to obtain HP fragments with well defined structures [14–17]. More recently, modular or combinatorial approaches have been devised to rationalise and speed up the synthesis of libraries of HS/HP fragments displaying various sulfation and epimerisation patterns [18,19]. Such synthetic approaches being extensively described in recent reviews [12,20–22] we will not describe further on in this review such syntheses of HS and HP hypervariable fragments.
3 SAS charge topologies: a second molecular diversity level
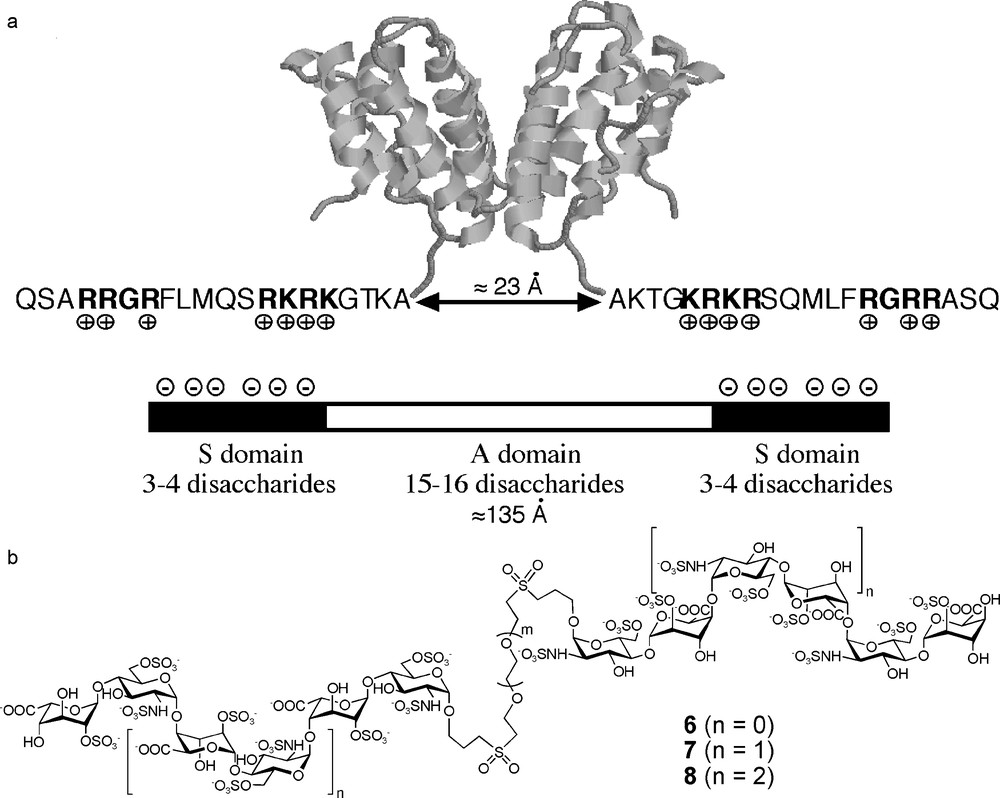
One and a half decades ago, it appeared that the existence of regulated hypervariable domains in HS could not account for all the regulatory effects of this polysaccharide and that many interactions appear to depend more on the overall organization of HS domains than on their fine structure [23]. Indeed, the microheterogeneities resulting from variation in sulfation and epimerisation patterns represent only a first level of molecular diversity in HS (Fig. 3a). In fact, the polymer, typically composed of 50–200 disaccharide units (25–100 kD), is not fully heterogeneous. Quite regular N-acetylated regions (A-domains), mainly composed of d-glucuronic acid and N-acetylated glucosamine, and thus with low global charge, separate domains rich in l-iduronic acid and N-sulfated glucosamine (S-domains) which are hypervariable and highly charged (Fig. 3a). The latter are typically three to eight disaccharides long while the A-domains are more regular and encompass a larger area around 15 disaccharides in length [24]. In between, mixed A/S-regions of variable length make the transition between A and S-domains. Thus, in addition to the first level of molecular diversity discussed above, HS presents a second level of diversity due to the various combinations of S, A and A/S-domains generating multiple SAS charge topologies along the polymer chain (Fig. 3b).
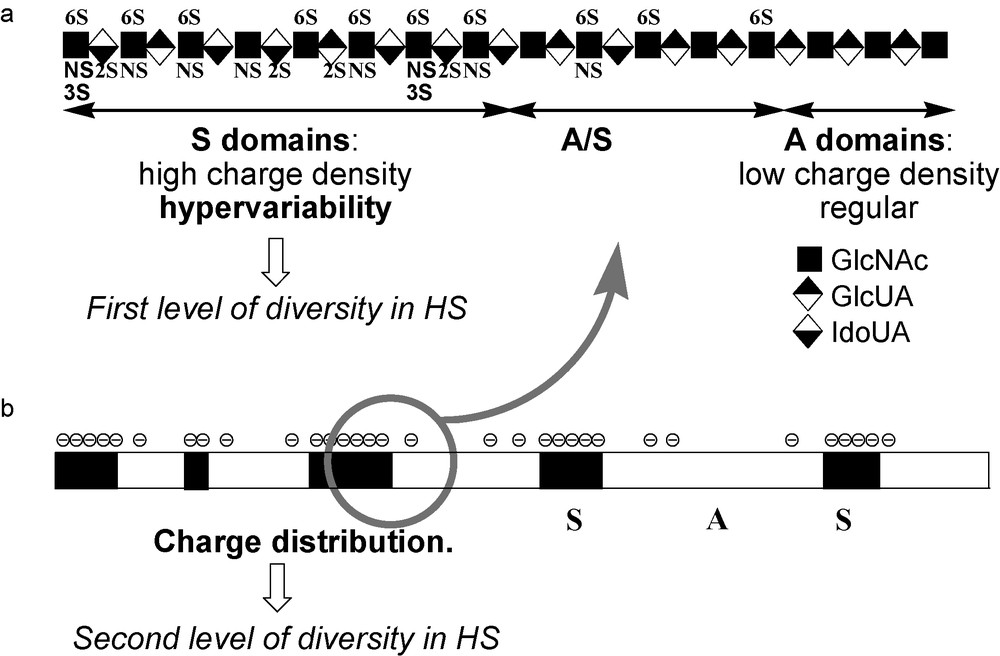
Dual molecular diversity in HS chains. a) Hypervariability resulting from the various epimerisation and sulfation pattern generate a first level of molecular diversity. b) The charge distribution due to the alternation of S and A-A/S-domains of variable length, leads to different SAS charge topologies that generate a second level of molecular diversity.
The primary interaction between HS and a protein is an attraction between the highly negatively charged S-domains and clusters of basic residues at the protein surface, mainly arginines and lysines. In some cases, for example, with AT-III [12,13], FGFs [15], or SDF-1α [25], a single S-domain is sufficient to allow a high affinity interaction, whose specificity is then linked to the uronic acid and sulfation pattern of the S-domain. However, with other proteins such as IFN-γ [23], PF-4 [26], RANTES(9-68) [27] or MIP-1α [28] a single S-domain is too short for high affinity binding and a longer fragment, including an A-domain, is needed for an efficient interaction. In fact, the above-cited proteins are either multimeric in solution, or multimerise upon binding to HS and the requirement for two distant S-domains reflects the fact that at least two basic domains of different subunits have to interact with the HS polymer. If the HS chains expressed at the cell surface display SAS domains whose charge topology is complementary to that of the basic clusters of the protein, a high affinity binding may occur (Fig. 4a). On the contrary, lower affinity are obtained if the polysaccharide and protein charge topologies do not match. Indeed, it has been proposed that the HS chain may adapt its conformation in order to meet the needs of recognition of a protein (Fig. 4b) [29]. However, this will have an enthalpic and entropic cost and will thus favour the binding of domains with the correct preformed geometry. In this regard, HS glycoconjugates, in which a linker of an appropriate length separates two S-domains, should be a selective functional mimetic of the binding site of a given protein on HS (Fig. 4c). Such considerations drove us when designing a general strategy for the preparation of libraries of HS SAS long fragments mimetics (section 4.2) [30].
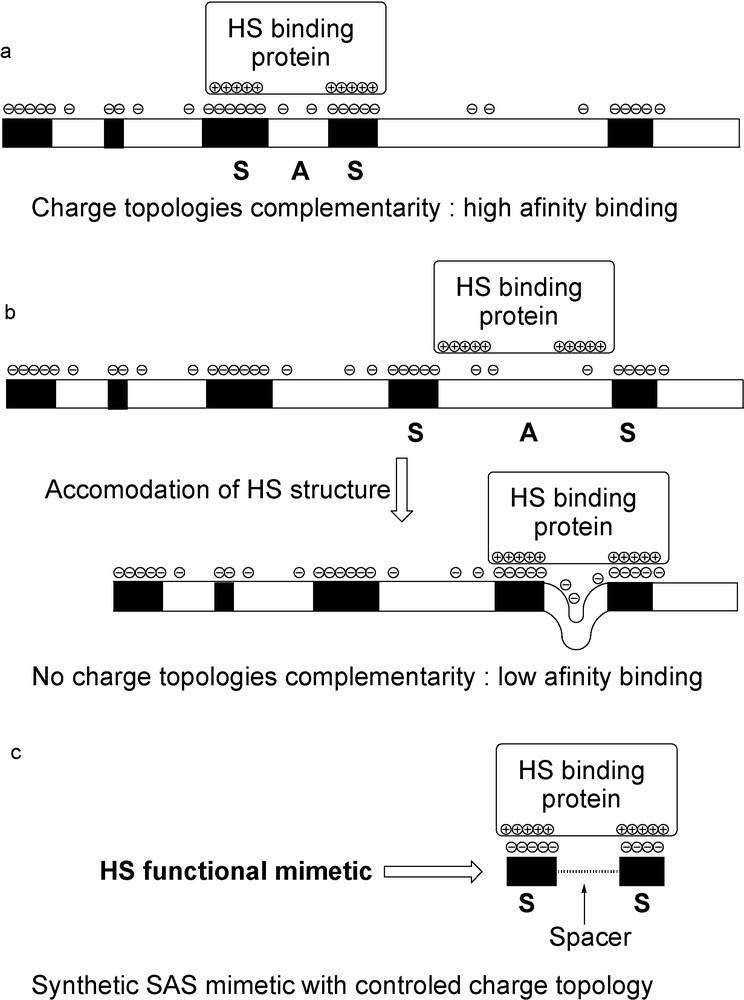
Model for the binding on HS polymer of a protein needing more than a single S-domain. a) High affinity when charge topologies are matching. b) Accommodation of the HS polymer conformation to meet the needs of a HS-binding protein: low affinity binding. c) Two S-domains linked by a spacer of optimum length should represent a functional mimetic of the SAS binding site and lead to high affinity binding constants.
4 Synthesis of HS and HP long fragment mimetics
In the mid 1990s, as it appeared that “small” hypervariable domains of HS and HP could not account for all the biological roles of these polymers, it occurred to glycochemists that the synthesis of long fragments was the next frontier to reach in order to prepare new highly bioactive and selective compounds. However, even taking into account the remarkable progress performed in HS and HP fragment synthesis cited above, synthetic carbohydrate chemistry is not yet mature enough to allow the preparation of HS/HP fragments as long as the 40- to 50-mer needed for efficient binding of some proteins [31]. Approaches in which a portion of the long fragment is replaced by non-carbohydrate or non HS/HP type carbohydrate spacers have thus been devised and are described below.
4.1 Synthetic anti-thrombotics with the full activities of heparin
Heparin catalyses the inhibition of two key proteases in the blood coagulation cascade: factor-Xa and thrombin (factor-IIa). As described above, an efficient inhibition of factor-Xa requires the binding of a specific pentasaccharide onto AT-III. The interaction of the pentasaccharide induces a conformational change in AT-III, which accelerates the inhibition of factor-Xa, but not of factor-IIa. Inhibition of the latter requires AT-III and factor-IIa to bind the same heparin chain in a ternary complex. Based on crystallographic studies, a model of this complex has been built [32], showing that the Thrombin Binding Domain (TBD) of heparin should be located at the non-reducing terminus of a heparin fragment in which an AT-III specific pentasaccharide would be located at the reducing end. Moreover, this model also show that heparin forms a bridge between two positively charged regions of the IIa-AT-III dimer, and that this bridge, which is approximately eight sugar in length, does not interact with positively charged residues. These considerations inspired the design of different types of glycoconjugates that should display AT-III mediated inhibition of both factors Xa and IIa. The general strategies implied the tethering of a fully sulfated maltotriose moiety as TBD (factor-IIa does not need a specific sequence for efficient binding), to an Antithrobin Binding Domain (ABD). For the later idraparinux 2 structure (currently in Phase III clinical trials, Fig. 5) was chosen, instead of the natural fondaparinux 1, for its ease of synthesis, its higher affinity for AT-III and better pharmacokinetic properties in vivo [12]. Following this general design, two tethering strategies were developed, the first one involving the use of flexible non-carbohydrate spacers [32–33] and the second one more rigid carbohydrate ones [34–36]. This impressive amount of work, summarised in a seminal review [37], allowed the synthesis of a great numbers of glycoconjugates in order to ascertain the general organisation of the long fragment mimetic (i.e., TBD at the non-reducing end and ABD at the reducing one, Fig. 5) and to determine the optimal length of the spacer to reach high anti-IIa and anti-Xa activities.
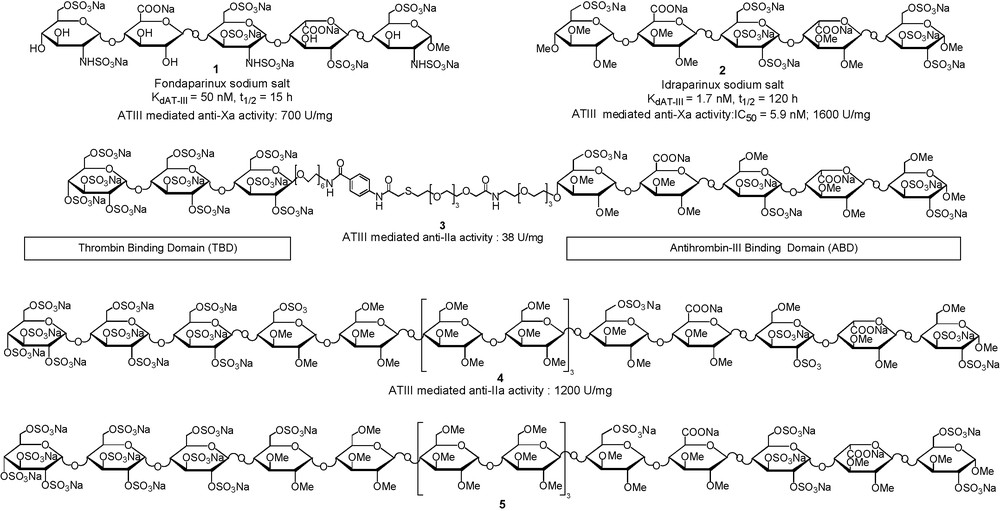
Comparison of Fondaparinux 1 and Idraparinux 2 structures and selection of tailor-made “synthetic thrombin-inhibiting heparin mimetic without side effects”.
As shown in Fig. 5, the anti-IIa activity of compound 3, containing a flexible non-carbohydrate linker is 30 times less than that of compound 4, which already displays an activity superior to heparin. The rigidity of the carbohydrate type spacer, and thus its preorganisation, probably lower the entropic cost due to the organisation of the flexible linker in 3, when forming the ternary complex with factor-IIa and AT-III. Moreover, the use of a non-charged trimaltoside spacing region, which was suggested by the fact that the bridge between factor-IIa and AT-III binding domains does not interact with positively charged residues, resulted in the preparation of molecules much less charged than heparin. This represents a major advantage since such compounds are unable to form the immunogenic PF4-heparin complex responsible for heparin-induced thrombocytopenia, a dangerous side effect of heparin. After structure–activity relationship studies, the 16-mer 5 was selected for pharmaceutical development of the first “synthetic IIa-inhibiting heparin mimetic without side effects”.
4.2 Synthesis of tailor-made glycoconjugate mimetics of HS that bind IFN-γ in the nanomolar range
In order to address the question of the second level of molecular diversity found in HS chains, we developed in Orsay a strategy that opens up easy access to libraries of HS functional mimics by combining HS synthetic fragments and α,ω-bis-thio-polyethylenglycols (PEG) of different lengths (Fig. 6b) [30].
Glycoconjugates as mimics of the IFN-γ HS binding site.
The binding of IFNγ to HS has been characterised some years ago [31] and was found to control the blood clearance [38] and the subsequent tissue targeting, accumulation, and localization of the cytokine [39]. Thus, the discovery of compounds able to modulate the activity of IFN-γ, by competing its binding on cell surface HS, could open the way to new immunotherapies [40], especially in the field of Crohn's disease and ankylosing spondylarthropathies [41]. IFN-γ is a C2 symmetric homodimer in solution and binds to HS by virtue of basic residues located at the C-terminus of the two subunits (Fig. 7a) [42]. A fragment of HS polymer, retaining the activity of the full-length polysaccharide, has been isolated using a protection approach (an enzymatic digestion of HS polymer in the presence of IFN-γ) [23]. The biochemical analysis of this fragment revealed that it was composed of two small hexa- to octasaccharide S-domains, separated by a more extended A-domain of 15–16 disaccharides (130–140 Å [43], Fig. 6a). A model of the interaction between IFN-γ and HS was thus proposed in which the KRKR domains [42] located at the C-terminus of each subunit of the IFN-γ dimer would interact with the highly charged S-domains of the fragment [23]. In order to confirm this hypothesis and to obtain compounds able to bind selectively and with high affinity to IFN-γ, we decided to prepare glycoconjugates 6–8, in which two S-domains, ranging from tetra- to octasaccharides, would be linked by their reducing end to a PEG based linker (Fig. 6b). We chose PEG linkers for their water solubility and their availability in different lengths. As discussed above, the use of flexible linkers may lead to lower binding constants with respect to more rigid spacers. However, such a flexibility may be important to mimic A-domains whose conformational plasticity is thought to be important for their biological role [29]. Moreover, PEGs are essentially non immunogenic and their incorporation into a drug is not a problem from the pharmacological point of view.
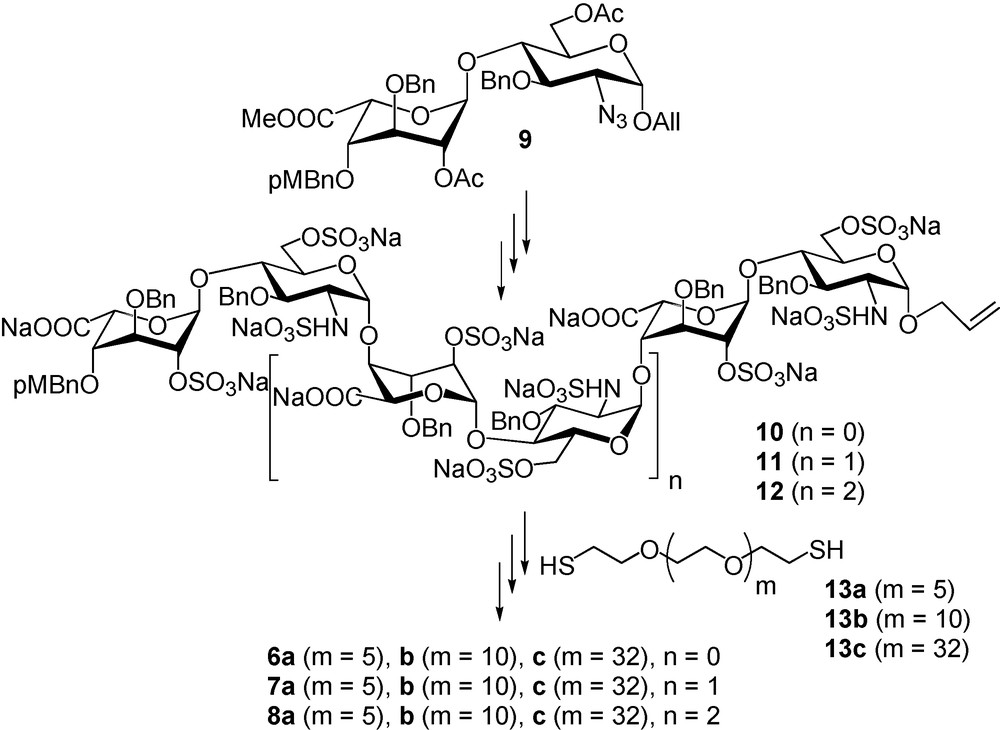
A library of SAS mimetics.
Disaccharide 9 [44–47] was successfully used as a starting building block for the preparation of sulfated, but still benzylated and allylated, tetra, hexa and octasaccharides 10–12. These compounds were conjugated onto α,ω-bis-thio-PEGs 13a–c, via UV promoted thiol-ene coupling, to give benzylated glycoconjugates that were hydrogenolysed after oxidation of the thioether function into sulfones (Fig. 7). The nine glycoconjugates 6a–c, 7a–c and 8a–c, as well as the corresponding non conjugated tetra-, hexa- and octasaccharides were then tested for their ability to inhibit IFN-γ/HP interactions using surface plasmon resonance. Preliminary experiments demonstrated that among the nine glycoconjugates, only 8a–c strongly inhibits the IFN-γ/HP interaction (IC50 of # 35 nM, that is in the range of the Kd of IFN-γ on HS), while the inhibition by the sole octasaccharide is low. Moreover, there is a clear dependence on the activity with the linker length. Compound 8b (50 Å linker length) is the most potent inhibitor while 8c (114 Å linker length) is the less active, compound 8a (33 Å linker length) having a medium activity (Fig. 8a). Since the C-terminus of IFN-γ is implicated in the cytokine binding to its receptor [48], the ability of glycoconjugates 6–8 to inhibit IFN-γ/IFN-γ-receptor interaction was also studied [49]. For that purpose, we immobilised the ectodomain of the IFN-γ-receptor on a Biacore chip and analysed the ability of the synthetic glycoconjugates to inhibit the IFN-γ/IFN-γ-receptor interactions. We thus found that the inhibitory activity of the glycoconjugates in this system closely parallel their ability to inhibit IFN-γ/HP interactions [49]. In view of these data, we investigated whether 8b would be able to downregulate the pro-inflammatory activity of IFN-γ in a cellular model. To this end, we measured the ability of this glycoconjugate to prevent the induction of the HLA-DR antigen by IFN-γ on Colo 205 cells. Our data (Fig. 8b) showed that 8b clearly displayed anti-IFN-γ activity, with an IC50 close to 1.5 μg/mL (250 nM).
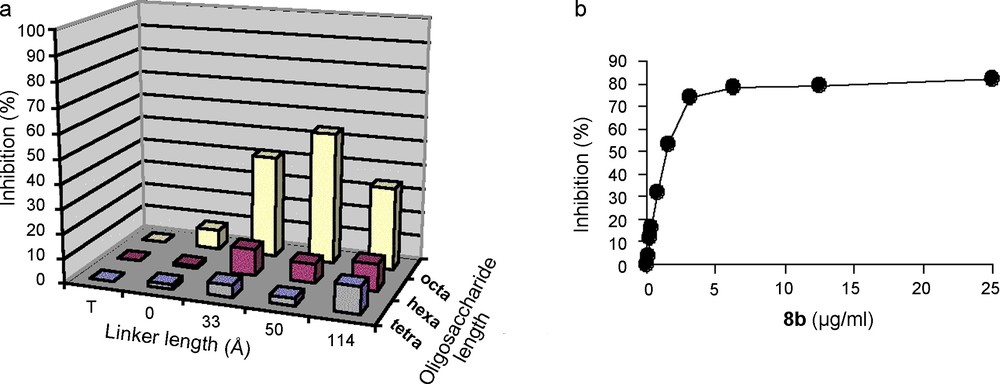
a) IFN-γ/HP binding inhibition by synthetic glycoconjugates (T: buffer, 0: non-conjugated tetra, hexa- and octasaccharides). b) Dose dependent inhibition of the expression of HLA-DR molecules at the surface of Colo 205 cells following IFN-γ stimulation.
We have thus demonstrated that glycoconjugates in which S-domains are separated by PEG linkers are good tools to access the “second level of diversity” found in HS chains. The SAS mimetic library 6–8 constitutes a valuable set to decipher the general charge organisation needed for a protein to bind efficiently a HS SAS domain. Regarding IFN-γ, the glycoconjugate 8b efficiently mimicking the HS binding site and this cytokine is thus able to inhibit both IFN-γ/HP and IFN-γ/IFN-γ-receptor interactions with an IC50 of # 35 nM. Consequently, 8b is able to inhibit one of the pro-inflammatory roles of IFN-γ. Based on this scaffold, our future work will focus on determining if the HS microheterogneity pattern(s) specific for IFN-γ can be identified, in order to enhance the specificity 8b, and on investigating whether such type of molecules could be of interest in pathologies for which IFN-γ has been identified as a target.
5 From HS and HP long fragment mimetic to multivalent chimeras
As shown above, tailor-made glycoconjugates mimicking a HP or a HS long fragment are invaluable tools to characterise the structural determinants, allowing a template effect or a cooperative binding to occur. However, mimicking natural HS or HP fragments was only a first step towards the preparation of highly bioactive drug hits to drug candidates as it occurred to glycochemists that combining an HS/HP fragment to another bioactive molecule could lead to chimeras having a multimodal activity.
5.1 From long fragment mimetics to multimodal glycoconjugates
5.1.1 New HP-activity like conjugates with unprecedented pharmacological profile
HP, as well as Low Molecular Weight Heparins, are still the reference as antithrombotic drugs. However, one of their shortcomings is their inability to inhibit clot-bound factor-IIa which is the cause of rebound thrombin generation after successful thrombolysis. Fondaparinux 1, idraparinux 2 do not circumvent this imitation and full length HP mimetics 3–5 will not. Inhibition of clot/thrombus associated thrombin, as well as the prevention of the formation of such complexes, may be achieved with direct factor-IIa inhibitors (DTIs). Unfortunately, the short pharmacokinetic of the DTIs tested preclude their use for prolonged treatment. Based on data showing that the half-life of an AT-III binding pentasaccharide is governed by its affinity for this protein, conjugates in which an idraparinux derived ABD (half-life: 120 h in man) would be linked to a DTI, in order to modulate its half-life, were designed. The NAPAP derivative 16 (Fig. 9) was first chosen as DTI for its significant anti-IIa activity and the possibility to functionalise the α-carbon of its glycin moiety without loss of activity [50]. The oversulfated version of idraparinux, compound 15, was selected as ABD for its high anti-Xa activity and affinity for AT-III. The resulting conjugate 14 displayed a unique antithrombotic profile in vitro: a high anti-IIa activity (IC50 = 0.35 μM) as well as AT-III mediated anti-Xa activity (885 U/mg). In vivo, the measurement of the antithrombotic activity in an aorta-flow model revealed that conjugate 14 is a stronger inhibitor than a combination of 15 and 16 [50]. Moreover, pharmacokinetic studies indicated that the conjugate 14 has a half-life of 1.5 h in rat versus 9 min for 16 alone, confirming that the half-life of this novel class of antithrombotic is determined by the interaction of its ABD with AT-III. In order to allow further pharmacological developments, an optimisation of the NAPAP derivative 16 was performed. A 40-fold increase in the anti-IIa activity was obtained when replacing the naphtyl moiety by a 4-methoxy-2,3,5-trimethyl-phenyl (compound 18, Fig. 9), allowing a better matching of the anti-IIa and anti-Xa activities of the two moieties of the chimera 17: IC50 = 15.0 nM in the inhibition of factor-IIa by the DTI moiety 18 and IC50 = 5.9 nM for the AT-III mediated inhibition of factor-Xa by idraparinux 2 (Fig. 5). The resulting conjugate 17, whose pharmacological profile is as promising as that of 14 was selected for further development [51]. In vitro, this candidate has several advantages over existing antithrombotic drugs: it inhibits factor-Xa in the presence of AT-III, it has a direct AT-III independent effect on both free fluid and clot-bound factor-IIa, it exerts a dose dependent effect on many coagulation tests thanks to the DTI moiety, it inhibits factor-IIa induced platelet aggregation and does not cross react with PF4-antibodies. In addition, promising results were obtained in vivo: a 100% bioavailability after subcutaneous administration, a half-life in rat of 3 hours (10 times higher than unconjugated 18) from which a half-life in man of 30–36 h can be estimated. Moreover, the glycoconjugate 17 shows a unique ability to prevent thrombotic reocclusion as opposed to established treatments [51].
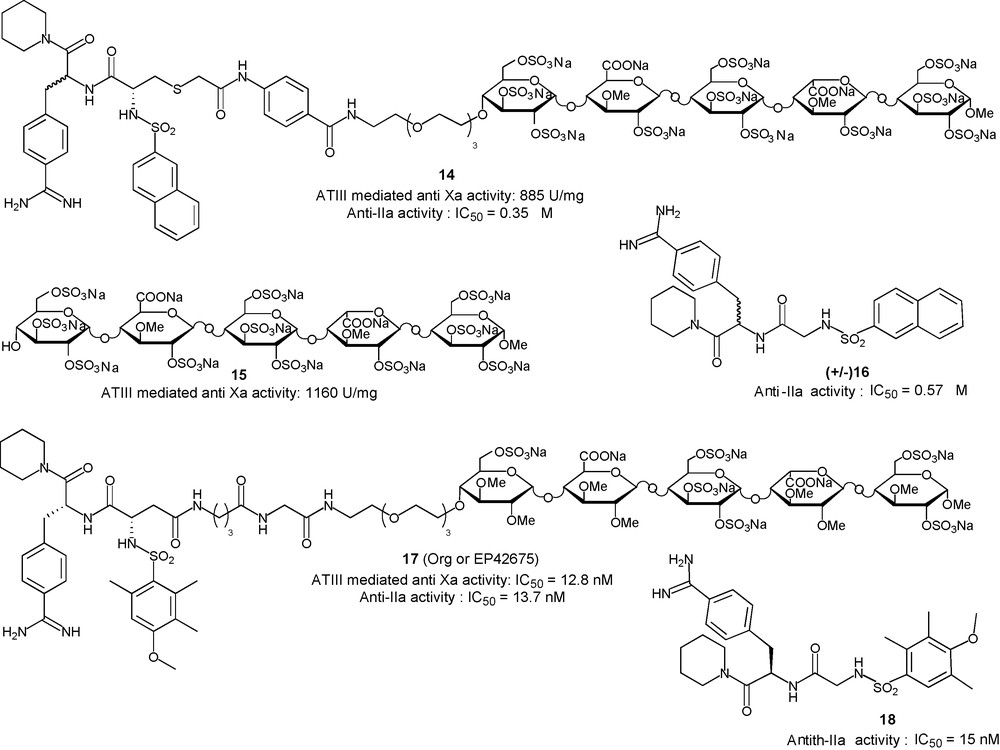
Glycoconjugates to inhibit factor-IIa in a non AT-III dependent way.
5.1.2 Introducing biotin: a third player for immediate shutdown of antithrombotic activity
Increasing the half-life of antithrombotic drugs in vivo has been achieved with the synthesis of non natural mimetics such as idraparinux 2, mimetic 5 and chimeras 14 and 17. However, if such improvements allow one to treat patients with one weekly dose, there are some acute clinical situations where the presence of a long acting antithrombotic in a patient circulation may be problematic. With such compounds, there is a medical need for a potent fast acting antidote. To this end, a biotin moiety has been grafted, with various length spacers, on an idraparinux like pentasaccharide to give conjugates 19a–c (Fig. 10) [52]. The pharmacokinetic and pharmacologic properties of compound 19b were determined in vitro, ex vivo and in vivo showing that the coupling of the biotin moiety has no effect on the antithrombotic properties of the idraparinux moiety of the conjugate. Moreover, injection of avidin, that binds to biotin with a Kd of 10−15 and can be injected safely in humans at doses from 10 to 100 mg, allowed immediate and complete abrogation of the anti-Xa activity [53]. A similar strategy can also be applied to allow the instantaneous shutdown of the anti-IIa and anti-Xa activity of HP long fragment mimetics. To this aim, a strategy allowing the introduction of a biotin moiety in the saccharidic spacer region of the HP long fragment mimetic was devised (Fig. 10). A key step was the introduction of a masked amino group, using potassium phtalimide nucleophilic displacement of a 6-O-tosylate group, in the reducing end glucose of compound 20 to give the nonasaccharide 22. Further elongation of the oligosaccharide chain both at the reducing and non reducing end of 22 and deprotection lead to an hexadecasaccharide in which an amino-group in position 6 of the 13th unit allowed the introduction of a biotin, to give, after deprotection and sulfatation, the biotinylated HP long fragment mimetics 24a–c (Fig. 10) [52].
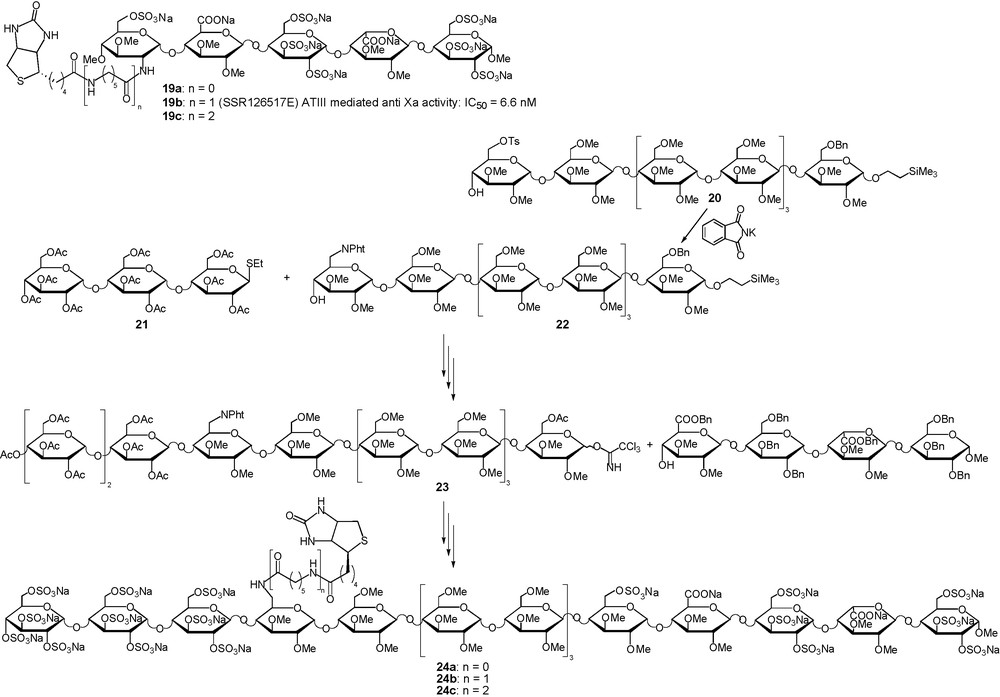
Neutralisable long half-life anticoagulant.
To make the idraparinux-NAPAP conjugate 17 an even more attractive anticoagulant drug particularly useful in acute clinical situations where there is a need for a potent fast-on/fast-off anticoagulant activity, a strategy was also devised for the introduction of a biotin on such conjugates. This “neutralizing moiety” was covalently linked to the spacer between the pentasaccharide ABD and the DTI parts to give the trimodal conjugate 25 (Fig. 11) [54]. As observed for the biotinylated version of idraparinux 19b, the introduction of the biotin moiety did not impair the activity of the conjugate 25 which was identical to that of 17. Gratifyingly, and in accordance with the wishes of the chemists who designed it, both anti-IIa and anti-Xa activities of 25 could be switched off instantaneously and simultaneously by injection of avidin [55].
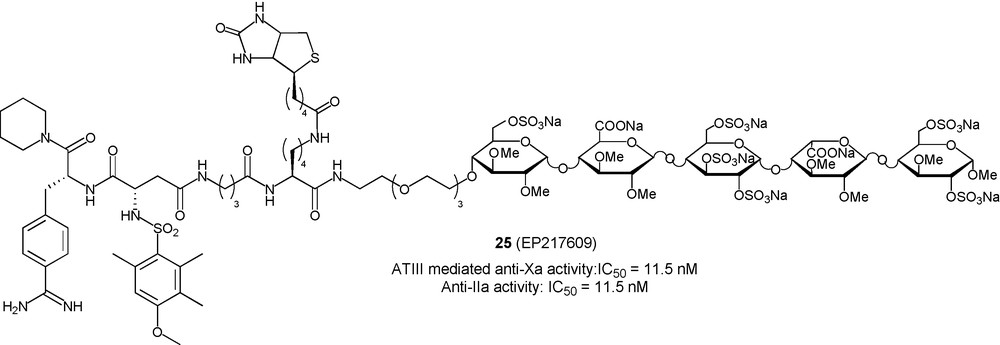
Introducing a biotin to switch off the antithrombotic activity of 17.
5.1.3 A synthetic CD4-HS glycoconjugate inhibit CCR5 and CXCR4 HIV-1 attachment and entry
Human Immunodeficiency Virus (HIV), the causative agent of AIDS has infected 60 million people worldwide and is one of the leading causes of death worldwide. Efficient drugs are available to inhibit HIV replication and to prevent the evolution of the infection to AIDS and death, but none of them is powerful enough to eradicate the virus. In addition, viral escape from drug control and side effects necessitate regular drug regiment changes. There is thus an urgent need for more powerful anti-HIV and identification of new therapeutic targets. In this regard, the blockade of viral entry is amongst the most promising recent approaches. HIV entry is initiated by the binding of the envelope glycoprotein gp120 to the primary receptor CD4 [56] which triggers conformational changes that relocate several domains of gp120 and fold a four stranded β-sheet known as the CD4-induced (CD4i) domain [57,58]. The structuration of the CD4i domain allows further interaction of gp120 with a coreceptor, usually either CCR5 or CXCR4 (Fig. 12a) [59,60]. This second interaction induces another conformational change of gp120 that triggers the penetration of fusogenic peptides of the gp41 into the cell membrane. Several further events result in cell-virus membrane fusion and entry of the viral capsid into the cell. HIV gp120 is known for a long time to bind to sulfated glycans and especially HS [61]. More recent data recent data have shown that the binding of gp120 on HS chains is greatly enhanced when the interaction is measured in the presence of soluble CD4 (sCD4), suggesting that the CD4i domain is an HS binding domain. Indeed, the reorganization of gp120 after binding to CD4 relocates, several basic amino acids in close proximity within the CD4i domain [62]. Based on these observations, it was hypothesized that a molecule composed of CD4 linked to HS would bind to gp120 through its CD4 moiety and expose the coreceptor binding domain, which would then become available to be targeted by HS (Fig. 12b) [63]. Such a molecule should simultaneously block the binding of gp120 to CD4, HS and coreceptors, and thus inhibit viral entry for both R5 and X4 viruses, that use respectively, the CCR5 and CXCR4 coreceptor to mediate their entry. A conjugation between recombinant sCD4 and an HS fragment isolated from natural sources could have allowed the preparation of such a glycoconjugate. However, since sCD4 is a large biomolecule and thus difficult to modify regioselectively to introduce a glycan chain, we thus opted for a total synthesis approach that allows preparing fully homogeneous and characterized glycoconjugates. In such an approach, a sCD4 surrogate, able to induce the folding of the CD4i domain, was designed based on the crystal structure of a scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 [64]. The 27-mer 27 (Fig. 13), whose sequence has been conceived in order to contain a single lysine residue, as future oligosaccharide attachment point, located in close proximity to the basic residues of the CD4i domain. This peptide was synthesized using Fmoc supported peptide synthesis, purified and folded. To our great delight, surface plasmon experiments were able to demonstrate that this peptide was able to bind efficiently to gp120 and to induce the folding of the CD4i domain, showing that it was an efficient CD4 functional mimetic [65]. In order to allow further conjugation of the HS moiety using a reliable methodology, a maleimido moiety was introduced on Lys5 to give the “activated” CD4 mimetic 28 (Fig. 13).
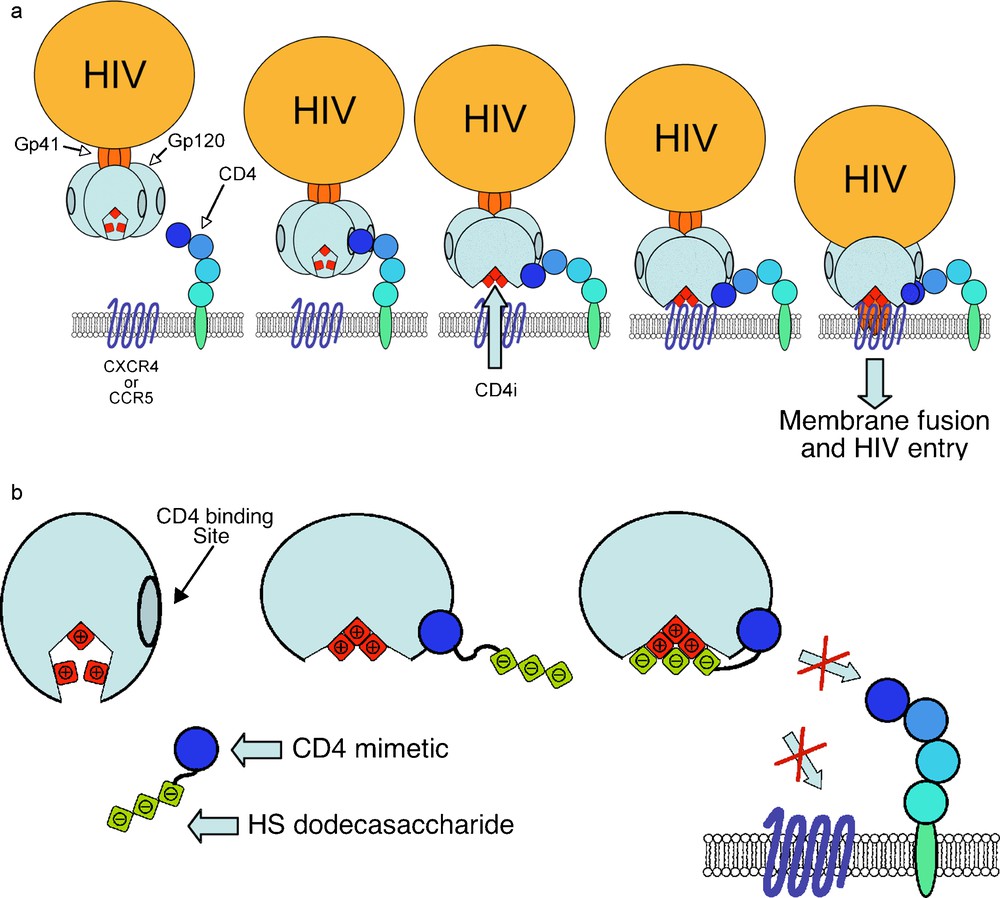
a) First steps of the HIV entry mechanism. b) A CD4-HS chimera to lock gp120 and block HIV interaction with its cell surface receptors.
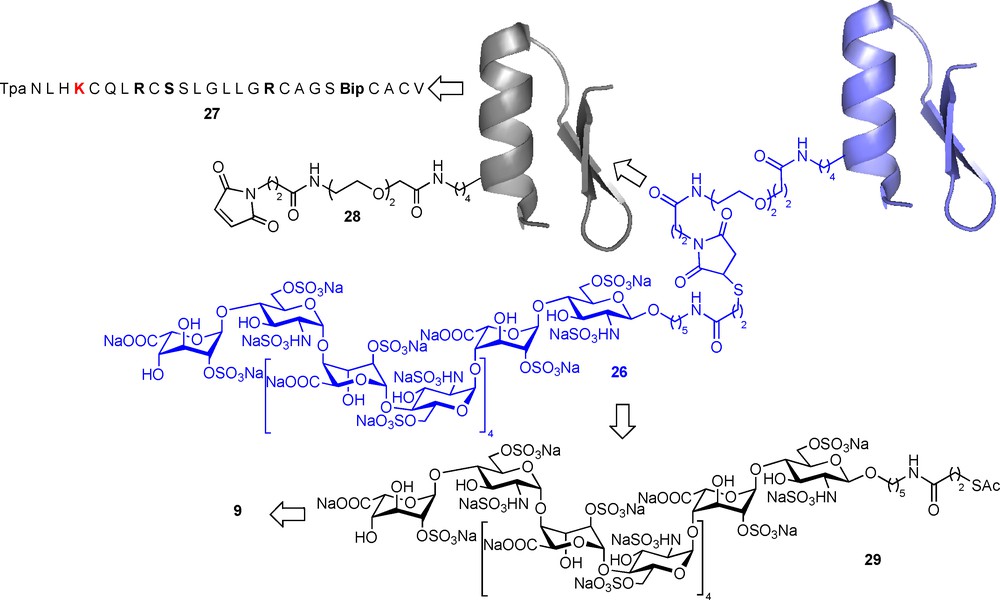
Total synthesis of a peptide-HS chimera to inhibit HIV entry.
For the synthesis of the HS dodecasaccharide moiety, we devised a highly convergent strategy, based on the well-documented efficient oligomerisation of the disaccharide building block 9 [30]. Indeed, after introducing a N-protected 5-aminopentanol moiety at the reducing end of this building block, the 2 + 2, 4 + 4, 4 + 8 elongation proceeded in high yields an full alpha selectivity as expected. The protected dodecasaccharide was then deprotected and sulphated as described [30], the reducing end amino group being deprotected during the methyl ester saponification step. A final reaction with SATP (N-Succinimidyl-S-acetylthiopropionate) gave the functionalised dodecasaccharide 29 (Fig. 13). In situ unmasking of the thiol functionality in 29 allowed further efficient conjugation to the maleimido activated CD4 mimetic 28 through Michaël addition [65]. The structure, as well as the high purity, of the resulting mCD4-HS12 conjugate 26 was confirmed by LC-MS analysis, confirming the pertinence of such a synthetic approach. The successful industrialization of the synthesis of hexadecasaccharides, such as 5, as well as peptides, such as the 36-mer fuzeon, indicates that such a hybrid molecule could be produced at larger scales for preclinical studies if needed.
Using surface plasmon experiments, we were able to demonstrate that mCD4-HS12 26, has the unique ability to fully and simultaneously inhibit, at a 1:1 molar ratio, the binding of both R5- and X4-gp120 to CD4 and monoclonal antibody 17b, used as coreceptor surrogate. As expected, a strong cooperative effect occurs between the peptide and glycan part of the molecule. Moreover, and not unexpectedly, mCD4-HS12 26 is also able to inhibit the binding of gp120 to HS chains. This is another interesting aspect of mCD4-HS12 activity since it has long been known that gp120 interacts with the widely expressed cell surface HS chains. This interaction is thought to allow HIV to stick on cells that do not express CD4 and to be presented afterward to more permissive cells. Inhibiting such interactions is obviously an added value for mCD4-HS12 with respect to other entry inhibitors that only blocks gp120 interaction with cell surface proteins. Having established the mechanism by which mCD4-HS12 interacts with gp120, we next investigated whether it displays antiviral activity. This was performed by measuring its ability to inhibit the infection of peripheral blood mononuclear cells (PBMCs) by the references strains HIV-1 R5 (Ba-L), dual tropic R5/X4 (89.6) or X4 (LAI). The replication of these strains was strongly and dose-dependently inhibited by mCD4-HS12. The effective doses giving 90% inhibition (ED90) ranged from 3 to 11 nM. HS12 itself had no activity in the concentration range tested, whereas the CD4 mimetic 27 had weaker activity (ED90 above 500 nM). Gratifyingly, none of these molecules showed cytotoxicity up to 1 μM. The mCD4-HS12 26 mediated inhibition of the infection of a pseudotyped virus with a VSV-G envelope, which enables viral particles to infect cells in a CD4 and coreceptor-independent manner, was next studied and proved irrevocably that the inhibition of HIV-1 replication induced by mCD4-HS12 26 relies on the blockade of the entry process by interacting with the HIV gp120 [65]. Further in vivo experiments will be necessary to demonstrate if the unravelling of this new HIV Achilles’ heel can give rise to new therapeutics.
5.2 Using the idraparinux-AT-III high affinity interaction to extend the pharmacokinetic profile of therapeutic proteins or peptides
Biomolecules such as hormones, antibodies or soluble receptors, either produced recombinantly or by synthesis, represent an increasing market. However, their use may be hampered by poor pharmacokinetics/dynamic (PK/PD) driving extensive research to develop methods to enhance the PK/PD profile. In this regard, the binding of a biomolecule to long-lived plasma proteins can be used with great benefit. One of those may be AT-III, which is present at high concentration in blood plasma (≈ 3 μM) and was unexplored until recently for such purposes. As detailed above in paragraph 5.1.3, the half-life of an AT-III binding pentasaccharide is governed by its affinity for this protein, it was thus tempting to test whether such a half-life could be transferred to a biomolecule covalently bound to a suitable AT-III binding pentasaccharide. As described below, this idea was tested on two proteins: insulin and fuzeon, giving rise to two other types of GAG-peptide chimeras.
5.2.1 Enhancing the half-life of insulin
Tight control over plasma glucose level in the treatment of insulin-dependent type-1 diabetes mellitus can be achieved by the injection of insulin. A natural overall physiological profile can be mimicked by the administration of short and long-acting insulin. Insulin is a dimeric protein in which the N-terminus of the B chain as well as its Lys29 are non essential for the bioactivity and can be considered as a good point of attachment for a glycan moiety. To test this hypothesis the two maleimido-activated insulin 33 and 34 were prepared as well as three thioacetyl functionalised idraparinux like pentasaccharides 30–32 (Fig. 14) [66]. Those three compounds differ in their total sulfate content, consequently their affinity for AT-III is different and should allow a fine-tuning of the conjugate affinity for AT-III. The four insulin-pentasaccharide conjugates 35–38 were prepared using thiol-maleimide conjugation chemistry after in situ liberation of the thiol moiety. After purification, ESI-MS and N-terminal sequencing allowed the full structural characterisation of the four chimeras [66].
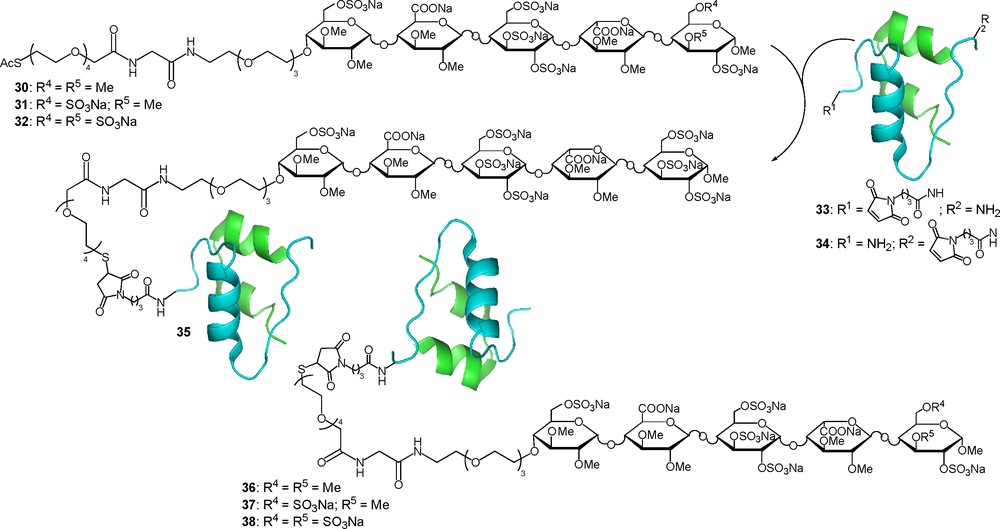
Enhancing the plasma half-life.
Before studying the PK/PD behaviour of conjugates 35–38, their binding affinity for AT-III was determined using surface plasmon resonance competition analysis in which the inhibition of the binding of AT-III onto immobilised 32 was determined. IC50 values of 96, 58 and 5.5 nM were respectively determined for 30, 31 and 32. Gratifyingly, the IC50 values of the conjugates fall in the same range, suggesting that their PK properties can be adjusted by changing the carrier pentasaccharide. This was indeed the case and the conjugates 35 and 38, binding the most tightly to AT-III, display an enormous enhancement of their residence time relative to unmodified insulin (t1/2 ≈ 5 h versus 10 min in rats). In parallel, plasma clearance and volume of distribution were also ameliorated. The ability of insulin glycoconjugates 35–38 to suppress glucose in non-diabetic rats in comparison with unmodified insulin was then determined. The increased exposure of all conjugates relative to insulin clearly gives rise to a longer duration of action that can be correlated to the observed differences in their ATIII-binding affinity and PK profile. Thus, suppression of glucose levels with the “long-acting” insulin glycoconjugates 35 and 38 lasted more than 7 h, whereas with unmodified insulin baseline levels were restored after 2 h. Moreover, in line with their decreased affinity for ATIII, conjugates 36 and 37 showed medium duration of action [66].
5.2.2 Enhancing the half-life of fuzeon
Enfuvirtide, also known as fuzeon or T-20, is a membrane fusion inhibitor approved for treatment against HIV. It is a 36-mer synthetic peptide, which prevents human immunodeficiency virus type 1 (HIV-1) from entering host cells. Despite its therapeutic efficacy, the clinical use of enfuvirtide is limited by a short plasma half-life (≈ 2 h), which requires twice-daily subcutaneous injections of 90 mg drug product. Improvement of the pharmacokinetic (PK) properties of this type of molecule is highly desired to facilitate its therapeutic use and to bring medical benefit to patients. To this effect, a strategy close to that described for insulin has been devised. As AT-III binding moiety, an idraparinux analogue was chosen that contains an unnatural 5-ethyl moiety in the glucuronic acid close to the reducing end and in which the amino group of a d-glucosamine unit at the non-reducing end is used as attachment point of the biomolecule [67]. Four different length PEG-containing spacers were introduced at this position to give the maleimido activated pentasaccharides 39-42. Further conjugation to the C-terminal cysteine modified fuzeon 43 gave the four chimeras 44–47 in which the distance between the antiviral moiety and the AT-III binding pentasaccharide is varied (Fig. 15).
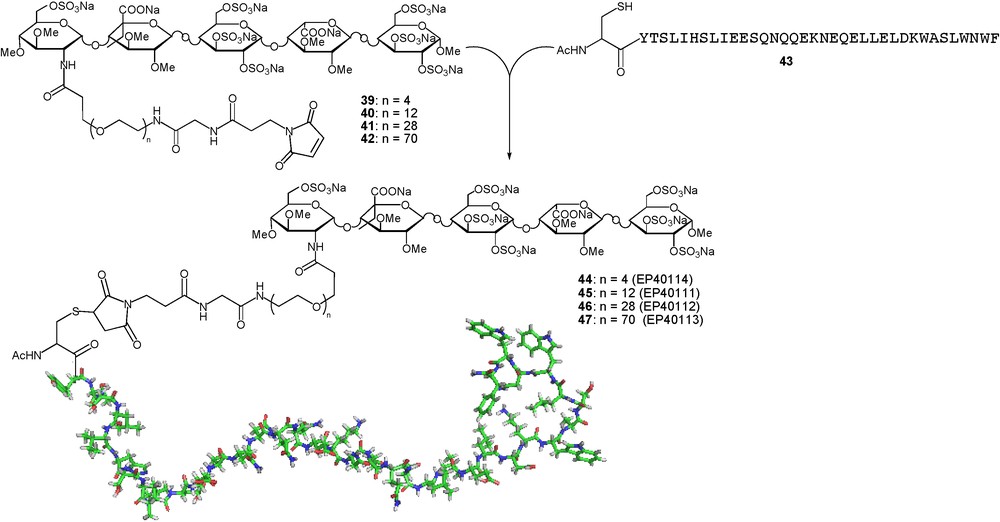
Enhancing insulin plasma half-life of fuzeon.
The affinities of the conjugates 44–47 for antithrombin were assessed by using an anti-factor-Xa bioassay, which is a reliable marker for the evaluation of the interaction between poly- and oligosaccharidic anticoagulants and AT-III. In this assay, the conjugates 44, 45, 46 and 47 inhibited the activity of factor Xa in a dose-dependent manner, with respective IC50 values of 93, 56, 53, and 32 nM, slightly lower than the activity of the pentasaccharide alone (IC50 ≈ 16 nM) but sufficient to hope for an efficient modulation of the PK profile of the conjugates. To test this hypothesis, the time-course profiles of conjugate 45 and fuzeon was determined in rat plasma after the administration of a single intravenous dose of the compounds at 4 mg/kg. Half-lives of 10.4 and 2.8 h were respectively measured for 45 and enfuvirtide. These data clearly demonstrate that the attachment of a carrier pentasaccharide to enfuvirtide significantly increased its plasma half-life [67].
The in vitro anti-HIV activity of the four conjugates was then determined in viral infection assays involving a CD4 T-cell line (CEM) and the reference HIV laboratory strains HIV-1 IIIB/LAI and HIV-2 ROD. All four enfuvirtide conjugates displayed potent anti-HIV-1 IIIB/LAI activity in the low-nanomolar range (EC50 ≈ 6 to 18 nM), comparable to the value for native T-20 (EC50 ≈ 3 nM). In these tests, compound 45 was shown to be consistently two- to threefold more active than the other conjugates. These values were comparable to the value for native enfuvirtide (EC50, 3 nM). Like native fuzeon, the conjugates were much less active against HIV-2 ROD (EC50 ≈ 280 to 990 nM). The ability of compounds 45 and 46 to inhibit the infections of PBMCs by different laboratory or clinical isolates of HIV X4 or R5 strains was then evaluated. When averaged, the mean EC50 of native fuzeon was about 20 nM, whereas the mean EC50 values of 45 and 46 were about 100 and 130 nM respectively [67]. In plasma, the conjugates will bind to antithrombin, which could putatively abolish their antiviral activity by steric hindrance. The effect of antithrombin on the antiviral activity of conjugates was thus studied under conditions mimicking the physiological situation in humans. The activities of conjugates 45, 46 and fuzeon in the inhibition of the infection of PBMCs with HIV-1 IIIB/LAI were determined the presence of 2.5 μM antithrombin. Both EC50 remained below 100 nM, but they were lowered by a factor 3 for 46 and a factor 6 for 45 [67].
The two examples reported in this paragraph demonstrate that the grafting of a tight AT-III binding pentasaccharide onto a biomolecule allows an efficient, tuneable and predictive enhancement of its PK/PD profile. The concomitant enhancement of the solubility of the polypeptide offers an added value regarding the bioavailability of the conjugate. An important issue in this approach is the initial biological activity of the biomolecule and to avoid diminution of its activity by sterical restriction once non-covalently bound to AT-III through the pentasaccharide moiety. It was indeed estimated that the glycoconjugate active concentration should stay below 50 nM to avoid any undesired AT-III mediated anticoagulant effect [66].
6 Conclusion
The chemistry of HS and HP has evolved from heroic and magisterial syntheses of the natural AT-III binding pentasaccharide sequence to the conception of mimetics and glycoconjugates with tailor-made activities. In this continuous evolution, tight collaboration between chemists, biologists, pharmacists and clinicians has been the driving force, allowing the various changes of paradigms observed in the field of HS/HP protein interactions. This evolution has also driven major methodology improvements in glycochemisty and the demonstration that the synthesis of up to hexadecasaccharide was amenable to successful industrial process development. Ongoing and future preclinical and clinical trials will demonstrate whether these new approaches can be turned into useful therapeutics for the benefit of patients.