1 Introduction
Glycosylation of proteins, the most diverse form of posttranslational modification, can play a key role in protein folding [1], and can crucially affect important protein properties [2] such as conformation and stability, susceptibility to proteases, and their circulatory lifetime. Moreover, glycosylation is also fundamental to many other key biological processes and is widely implicated in many aspects of cell–cell signalling, development, and immune response.
Protein glycosylation occurs most commonly at the amide nitrogen of particular asparagine (Asn) residues, so-called N-linked glycosylation. In the alternative, O-linked glycosylation, the attachment of glycans occurs through the side chain of serine or threonine residues. Amongst the many important mammalian proteins that are N-glycosylated, antibodies are of particular significance. For example, human IgG antibodies contain N-linked glycans at the two Asn-297 residues of the dimeric Fc (constant fragment) region and extensive work has confirmed that the precise identity of the glycans at these sites is key for modulating antibody-mediated responses, such as antibody dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [3]. Given that monoclonal antibodies (mAbs) are now widely accepted as the most important new class of therapeutic agents in development, the importance of control of N-linked glycosylation becomes obvious. N-Linked glycosylation can also be of particular significance for many mammalian enzymes. In the context of therapeutic agents, recombinant glycoproteins that have found utility as the basis of enzyme replacement therapies are of particular significance [4]. These increasingly important and widespread clinical applications have significantly increased the profile and importance of recombinant glycoproteins. However, since the biosynthesis of the glycan portions of these glycoconjugates is not under direct genetic control, then glycoproteins are produced intracellularly as heterogeneous mixtures of glycoforms; materials in which different oligosaccharide structures are linked to the same peptide chain. Cell expression systems almost invariably lead to the production of such inseparable heterogeneous mixtures of protein glycoforms. Access to pure single glycoforms of glycoproteins has therefore now become a major scientific objective [5]. Not only is it a prerequisite to facilitate more precise biological investigations into the different effects glycans have on protein properties, but it is also becoming an increasingly important commercial goal in the field of glycoprotein therapeutics [6], which are currently marketed as heterogeneous mixtures. Recently, the commercial and therapeutic significance of mAbs has added considerable financial impetus to the development of technologies that permit the production of defined glycoproteins in homogenous form. It is therefore not surprising that considerable effort has recently been expended in the attempted optimisation of the glycosylation patterns of mAbs. For example, the Biotech companies GlycoFi [7] and GlyCart [8] have focussed on cell engineering approaches to produce expression systems that can be used to produce mAbs with more ‘humanised’ glycosylation patterns. Similarly, other important glycoprotein therapeutics such as erythropoietin (EPO) have been the subject of glycosylation engineering in order to improve their therapeutic profile [9]. However, despite these apparent successes, it is important to remember that cellular approaches towards optimising the glycosylation profile of a recombinant glycoprotein actually give no guarantee as to complete glycoprotein homogeneity since they rely on cell biosynthesis.
On the other hand, in vitro access to entirely homogeneous single glycoforms of glycoproteins can be achieved by total synthesis of both glycan and polypeptide components, and several outstanding achievements in this area have very recently been published [10]. Although the application of techniques such as native chemical ligation does facilitate protein synthesis, the logistical challenges inherent in completing the total synthesis of both protein and associated glycans are still very considerable. Therefore, the more widespread application of such approaches will continue to require very considerable effort.
An alternative and attractive approach that may allow access to a wide range of glycoproteins bearing homogenous defined glycan structures, including mAbs, is that of in vitro glycoprotein remodelling (Fig. 1). The basic approach is as follows. In Step 1, the oligosaccharide chains of a heterogeneous mixture of glycoforms produced intracellularly are ‘trimmed’ back to a single GlcNAc residues at N-linked glycosylation sites using endohexosaminidase-catalysed hydrolysis. Endohexosaminidases (or more specifically endo-β-N-acetyl-glucosaminidases, ENGases) are a class of enzyme first identified in the early 1970s, the members of which specifically cleave the chitobiose core [GlcNAcβ(1-4) GlcNAc] of N-linked glycans between the two N-acetyl glucosamine residues. Since then endohexosaminidases, such as Endo H, have found widespread utility as biochemical tools to carry out this hydrolytic process [11], greatly facilitating protein manipulation and also allowing glycan profiling of the cleaved oligosaccharides. Following removal of the heterogeneous glycans, a single GlcNAc handle remains at each N-glycosylation site to which defined oligosaccharides may then be attached in a second and considerably more challenging step. In order to access natural N-glycan structures then, oligosaccharides must be attached to this GlcNAc handle en bloc. The only apparently practicable means of achieving such a daunting synthetic transformation is to once again use an endohexosaminidase, though this time to catalyse a transglycosylation reaction with a defined N-glycan Asn-linked oligosaccharide as the coupling partner. Studies in the 1990s revealed two endohexosaminidases that showed useful synthetic glycosylation activity in this respect: Endo M from Mucor Hiemalis [12] and Endo A from Arthrobacter protophormiae [13], both of which are members of the glycoside hydrolase (GH) superfamily 85 [14]. Seminal investigations revealed the remarkable ability (for some other leading references on synthetic applications of Endo A, see [15,16]) of these enzymes to effect transglycosylation of a variety of acceptor substrates bearing GlcNAc residues using full-length Asn-linked oligosaccharides as donors. However, since the enzymes naturally display hydrolytic activity, then the efficiency of these transglycosylation processes was quite limited; typically 100 to 1000 equivalents of glycosyl acceptor were required in order to effect even very moderate yields of product due to competitive hydrolytic reactions.
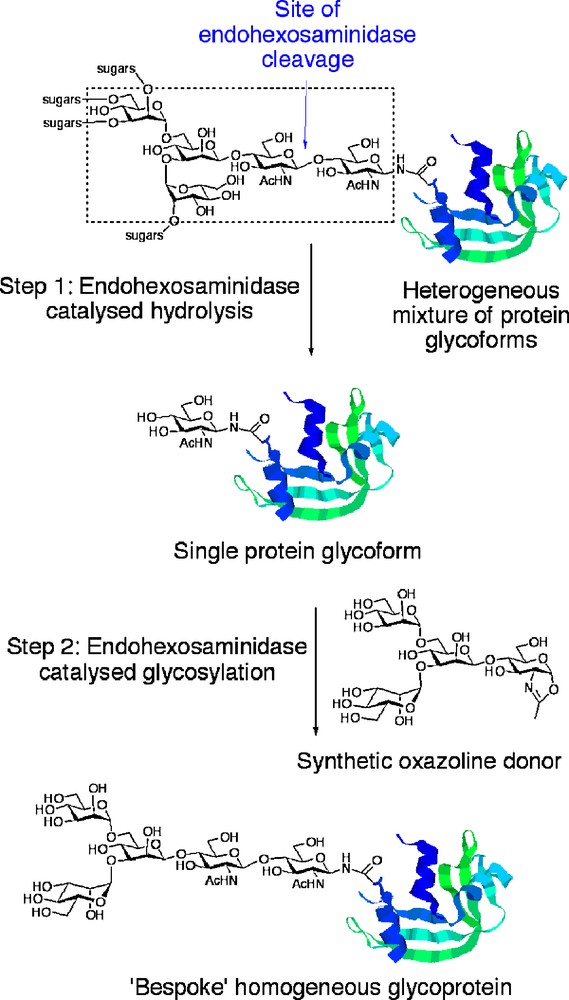
Glycoprotein remodelling with endohexosaminidases.
In 2001, a paradigm shift occurred when Shoda [17] and coworkers demonstrated that a disaccharide oxazoline could be used as an activated glycosyl donor substrate for these enzymes. This initial report immediately implied to us that other oxazolines may also be substrates for these enzymes, and furthermore that very substantial increases in synthetic efficiency should be possible using a whole range of oxazolines as activated donors. Thus, returning to the idea of glycoprotein remodelling, one could envisage a vastly more efficient Step 2 (Fig. 1) in which an N-glycan oxazoline is used as an activated donor. The 2001 Shoda report therefore further stimulated our interest [18] in the glycoprotein arena. In particular, the potential of chemical synthesis to access a whole range of oxazoline structures opened up new vistas of possibilities to us. However, the potential for future development was also not lost on other research groups, most notably that of Lai-Xi Wang. It later transpired that Wang and coworkers were also intent on developing synthetic methodology based on the initial Shoda disclosure in parallel with our work, although thankfully their studies and disclosures have largely been complimentary to our own [19].
In order to potentially extend and develop the applicability of the oxazoline approach two things were required. The first was access to a variety of N-glycan oxazolines, necessitating either total synthesis, or semi-synthesis using oligosaccharides derived from natural sources. The second was access to active endohexosaminidase biocatalysts that may be capable of transferring these oxazolines to GlcNAc residues attached to either peptides or proteins. This short review will centre on synthetic investigations undertaken by our group over the past seven years into the development of N-glycan oxazolines as activated donors for endohexosaminidase-catalysed glycosylation. The ultimate objective of the research program is, of course, the development of high yielding efficient methodology that will allow access to a wide variety of defined glycoconjugates, including glycoproteins and mAbs, in homogeneous form.
2 Chemical synthesis of N-glycan oxazolines
A primary objective was the establishment of robust synthetic routes to a variety of N-glycan oxazolines themselves for use in later biocatalytic studies. N-glycans may be obtained in small quantities from natural sources, such as ovalbumin, by degradation sequences, and the corresponding oxazolines can then be produced from them using semi-synthesis. However, such approaches are rather limited since only certain structures may be obtained using these methods, prohibiting both the establishment of complete structure activity relationships and the production of glycoconjugates bearing novel glycan structures. We therefore undertook to establish robust synthetic routes to structurally diverse N-glycan oxazolines employing a modular approach.
2.1 Natural glycans
Considerable synthetic endeavour has been expended on the synthesis of N-glycan oligosaccharides since they were first identified as biologically important and structurally challenging targets in the 1970s. However, the structures required for the biocatalytic experiments with endohexosaminidases require a single GlcNAc residue at the reducing terminus, rather than the natural chitobiose unit. Therefore, at the outset of this project, the vast majority of the requisite oligosaccharides and the advanced intermediates needed for their synthesis were novel materials, though routes were available which may be adapted to these particular targets.
The key consideration in N-glycan synthesis is formation of the β-manno linkage between what can be loosely termed the ‘central’ mannose and the GlcNAc units. The challenges inherent in the synthesis of any β-mannoside are well documented, and these are exacerbated when a poor acceptor such as the 4-hydroxyl of a protected glucosamine derivative is the partner for coupling. Indeed, our laboratory has specifically developed intramolecular glycosylation strategies for the synthesis of β-mannosides [20] based on the pioneering earlier work of the groups of Hindsgaul [21], Stork [22] and Ogawa [23], which have included the core N-glycan pentasaccharide. However, despite the attractiveness of intramolecular glycosylation strategies, in this instance, and for reasons that will become apparent after later discussion, we decided to adopt the C-2 epimerisation approach involving an SN2 reaction at C-2. There have been many variants and developments since the first disclosures of this approach in both its intramolecular [24] and intermolecular forms [25]. The most efficient and generally applicably procedure appears to be that reported by Schmidt [26] and Boons [27] in which Lev protection is used for the 2-position of a gluco thioglycoside donor such as 1. Glycosylation of donors 1 with the glucosamine acceptor 2 produced the desired β-disaccharides 3 in very high yield (Scheme 1) [18a,e]. The great utility of the 2-O-Lev protection is that Lev can be efficiently removed in a highly chemoselective fashion; something which did not prove possible for alternative ester protection of the 2-position due to competitive partial cleavage of the phthalimido protection of the glucosamine unit [18e]. Removal of the Lev with hydrazine acetate reveals the 2-hydroxyl which can be epimerised by a triflation/displacement sequence. In this latter respect, the report of the use of sonication by Fürstner [28] represents the key methodological development. Not only does the use of sonication greatly improve the reaction efficiency for a disaccharide substrate, to give a β-manno disaccharide such as 4, but it also means that the epimerisation sequence can be employed on/applied to considerably more complex oligosaccharides. For example, an alternative strategy was adopted for the synthesis of several N-glycan oxazolines in which the central mannose unit possessed a single mannose unit attached to the 3-position [18a]. Herein regioselective glycosylation of the 3-position of the gluco thioglycoside 5 with the manno trichloroacetimidate donor A (Fig. 2) and subsequent Lev protection of the 2-position gave rise to disaccharide 6. An analogous reaction sequence of glycosylation with glucosamine acceptor 1 to produce trisaccharide 7 and then Lev removal, triflation and acetate displacement with sonication gave the trisaccharide 8 in a highly efficient fashion [18a]. This latter example further demonstrates the utility of the Lev de-protection/triflation/displacement with sonication combination; indeed this epimerisation sequence has been successfully applied to substrates up to the pentasaccharide level.
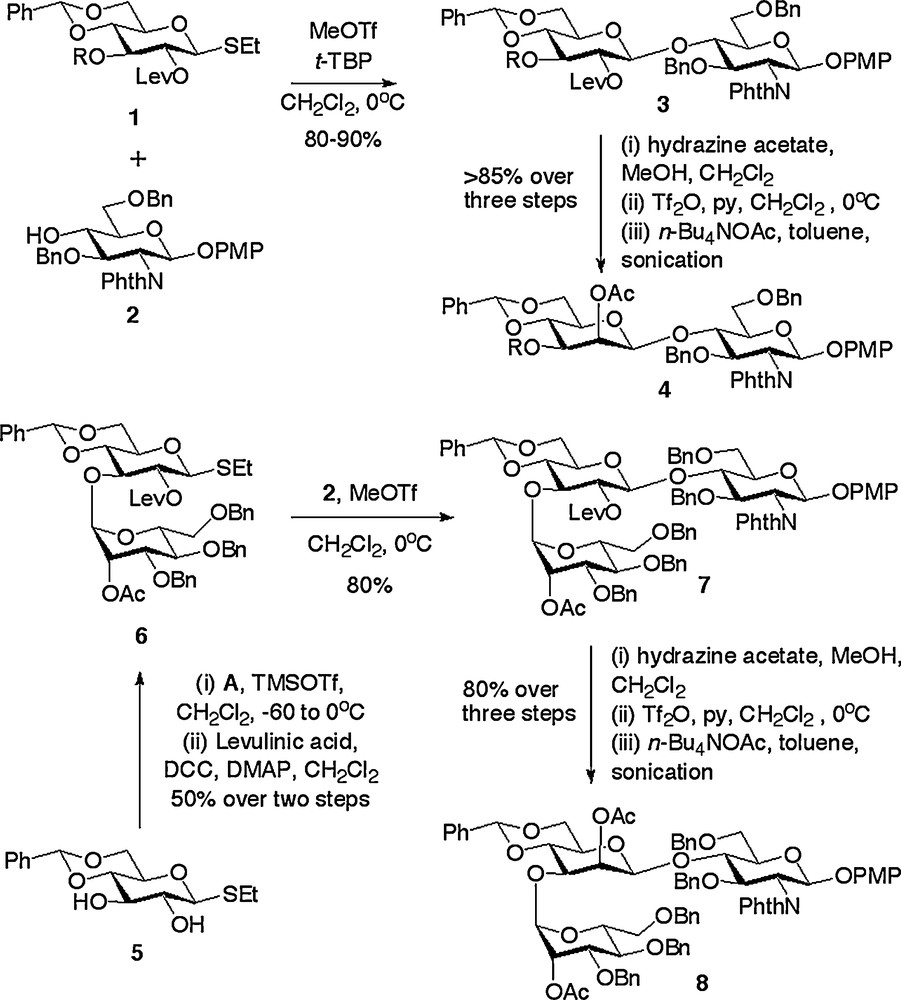
Synthesis of core building blocks via glucose to mannose epimerisation sequence.
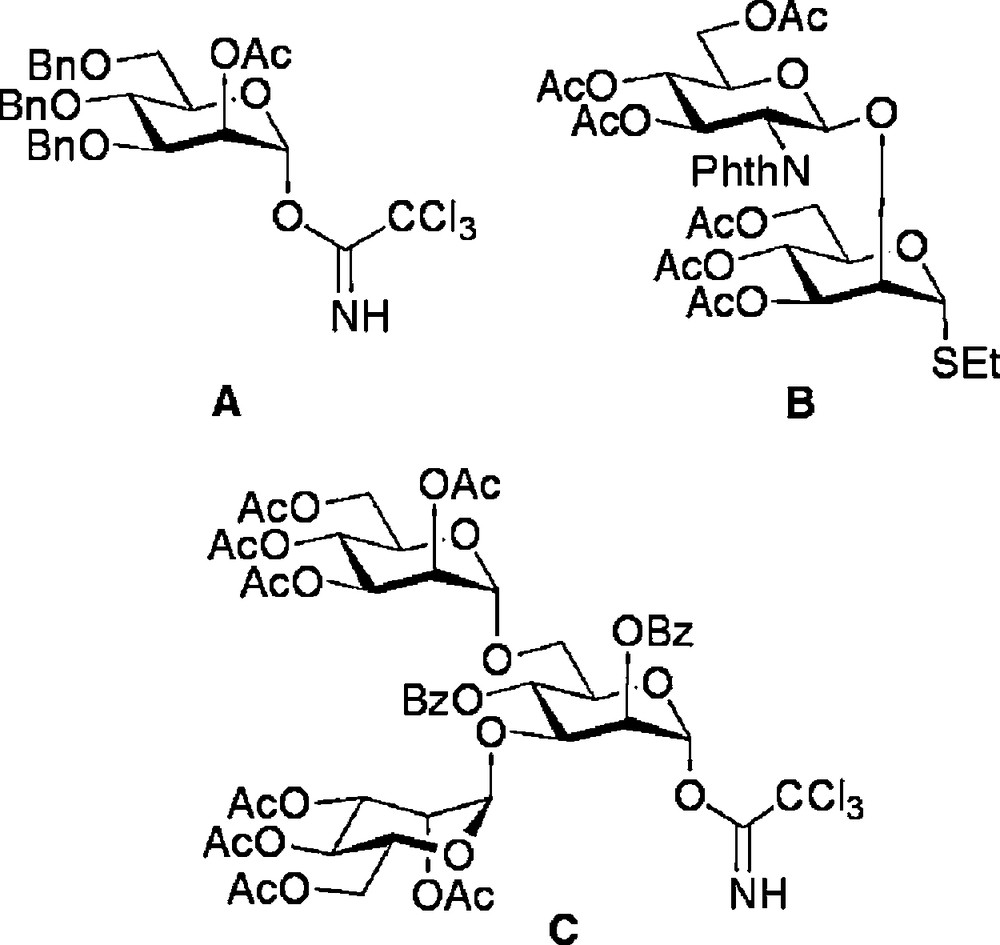
Glycosyl donors employed in modular N-glycan synthesis.
With routes to the core di- and trisaccharides established, decoration with additional structures by a modular approach was the most attractive strategy for the completion of the synthesis of a variety of N-glycan structures. Indeed, the power of modular construction has been elegantly demonstrated for the synthesis of full length natural N-glycans, most extensively by Unverzagt [29]. The glycosyl donors A, B and C (Fig. 2) were used for glycosylation of disaccharides 4 and trisaccharide 12 as shown in Schemes 2 and 3 to complete the oligosaccharide frameworks. Thus, regioselective ring opening of the 4,6-benzylidene of 4a revealed the primary hydroxyl group, which was then glycosylated with trichloracetimidate donor A to produce the (1 → 6) linked trisaccharide 9 [18c]. Alternatively, selective removal of the 3-O-allyl protecting group of 4b could be achieved by a two step sequence involving firstly isomerisation to the corresponding enol ether by treatment with a (1,5-cyclooctadiene)bis(methyldiphenylphosphine) iridium(I) hexafluorophosphate catalyst activated with hydrogen [30], and secondly, enol ether hydrolysis with N-iodosuccinimide (NIS) and water. Glycosylation of the free 3-hydroxyl with disaccharide donor B then gave tetrasaccharide 10. Regioselective ring opening of the 4,6-benzylidene of 10 revealed the 6-hydroxyl group which was then followed by a second glycosylation with disaccharide donor B to give the truncated complex N-glycan hexasaccharide 11 [18e].
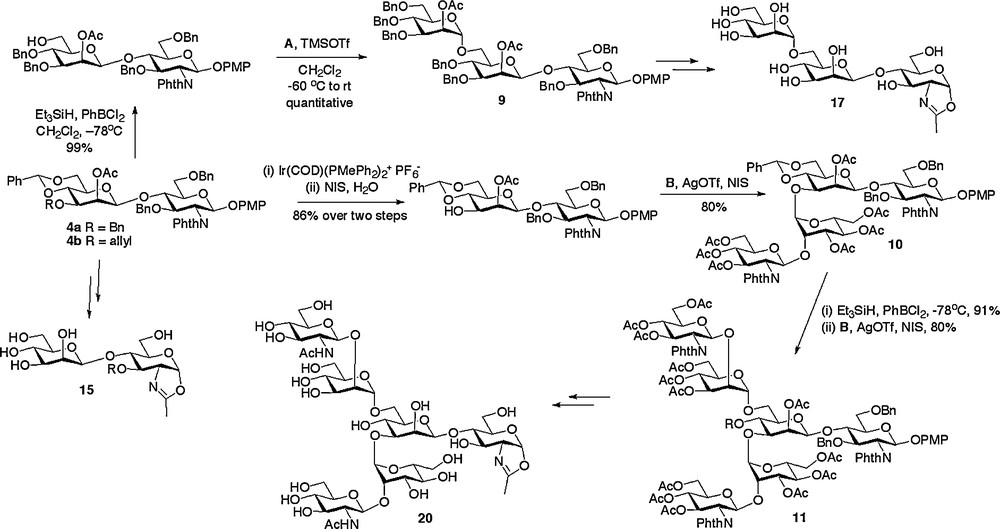
Synthesis of N-glycan oxazolines.
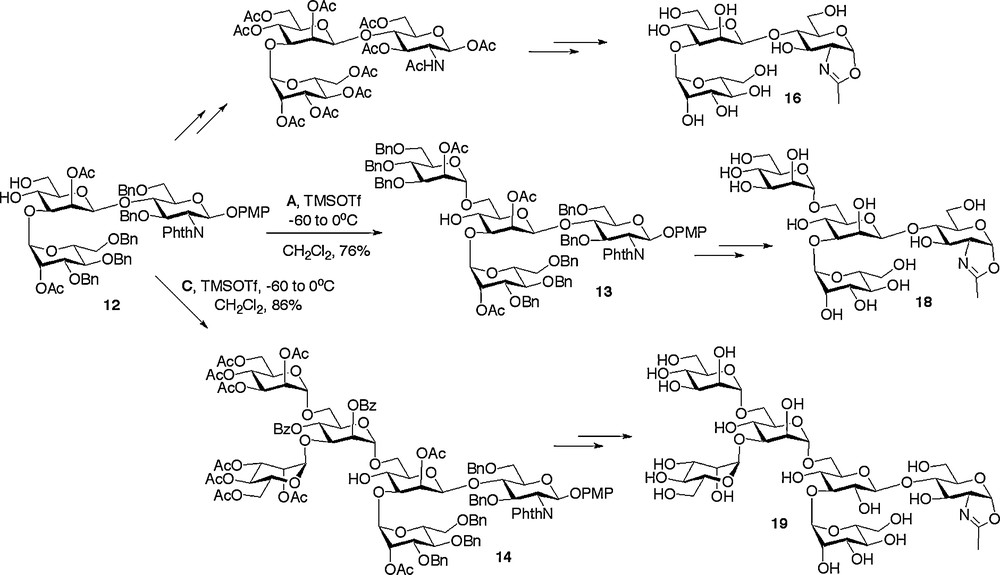
Synthesis of further N-glycan oxazolines.
Alternatively other high mannose structures could be accessed from the (1 → 3)-linked trisaccharide 12 (Scheme 3), which was itself formed by acid catalysed removal of the 4,6-benzylidene protecting group from trisaccharide 8. Regioselective glycosylation of the primary 6-hydroxyl with either monosaccharide donor A or trisaccharide donor C resulted in the high yielding synthesis of the tetrasaccharide 13 [18c] and the hexasaccharide 14 [18a], respectively.
With the assembly of the oligosaccharide frameworks completed, the final challenge was to convert them into their de-protected oxazolines. A prime consideration when considering the sequence of reactions to be employed was the acid sensitive nature of the final glycosyl oxazoline product. Thus a common sequence of protecting group manipulations was developed and applied generally. This sequence consisted of firstly conversion of all the alcohol protecting groups to acetates and the phthalamides to the required acetamides, then subsequent oxazoline formation, and finally global de-protection by de-acetylation under basic conditions. Oxazoline formation was certainly the most demanding of these synthetic conversions and a variety of methods were investigated. The optimum and most generally applicable procedure involved treatment of the peracetylated oligosaccharide with TMS bromide, BF3·OEt2, and tri-tert-butyl pyrimidine (t-TBP) in dichloroethane at 40oC for 6 hours; the use of the hindered non-nucleophilic base t-TBP instead of collidine led to a considerable improvement in the yield for this step. Application of this basic reaction sequence was applied to all available oligosaccharide frameworks, thus allowing the synthesis of disaccharide oxazoline 15, (1 → 3) and (1 → 6)-linked trisaccharide oxazolines 16 and 17, tetrasaccharide oxazoline 18, high mannose hexasaccharide oxazoline 19 and complex hexasaccharide oxazoline 20 (Schemes 2 and 3) [18a,c,e].
2.2 Non-natural glycans
The epimerisation strategy applied for the formation of the β-manno linkage detailed above, besides being high yielding, had another particular advantage. This was that simply omitting the epimerization step allowed us to follow analogous synthetic routes to access a complete array of N-glycan oxazoline analogues in which the central mannose unit was replaced by a glucose residue. Thus the non-natural N-glycans 21–24, depicted in Fig. 3, were readily synthesised [18b,c] following routes analogous to those depicted in Schemes 2 and 3, but by leaving out the gluco to manno epimerisation sequence. The particular desire to access non-natural glycans is explained in the next section.
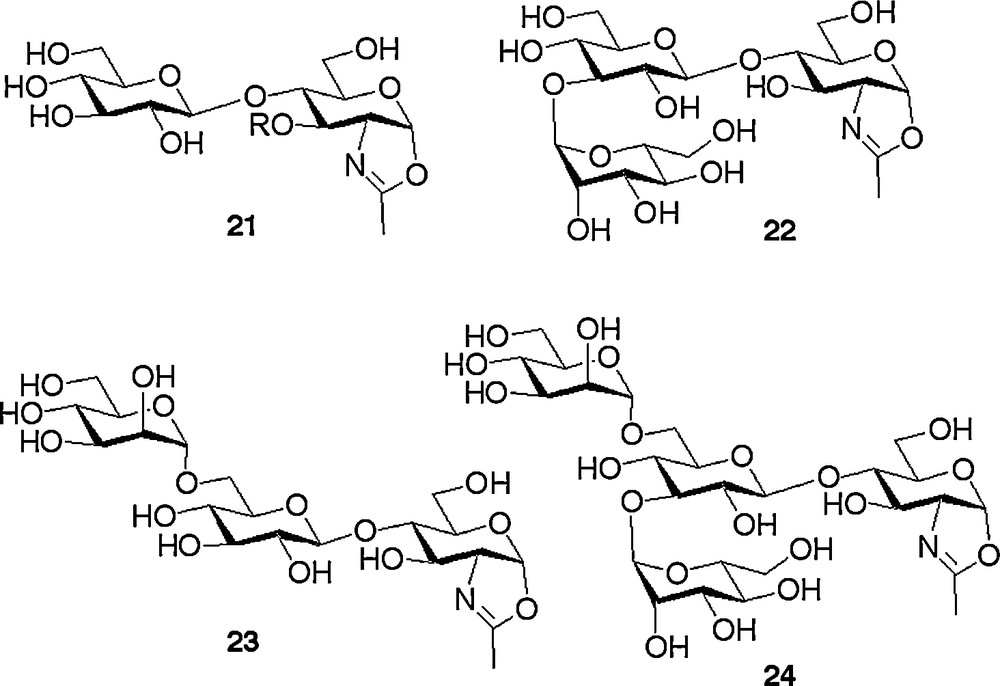
Non-natural oxazolines containing a central glucose residue synthesised by omitting the gluco → manno epimerisation step.
3 Glycosylation with endohexosaminidases
In order to investigate the synthetic efficiency of reactions catalysed by a variety of endohexosaminidase enzymes, a model acceptor was required which contained a UV chromophore so that the progress of synthetic reactions could be readily monitored by HPLC. The Z-protected glycosyl amino acid acceptor D (Scheme 4) appeared to be suitable, and was therefore synthesized from the corresponding GlcNAc glycosyl azide by a Staudinger ligation to a Z-protected aspartic acid derivative [18a]. The standard reaction used for investigation of the synthetic efficiency of endohexosaminidase-catalysed processes was therefore that shown in generalised form in Scheme 4. Importantly, the product of the synthetic reaction (rate constant k1) would usually be expected to be a hydrolytic substrate for the enzyme (rate constant k2). Minimisation of the rate of product hydrolysis relative to that of the synthetic reaction was considered to be key for the improvement of the overall synthetic efficiency of the process, for example by employing reaction conditions and using substrates for which k1 ≫ k2.

Endohexosaminidase-catalysed synthesis and competitive product hydrolysis using N-glycan oxazolines as donors with a model glycosyl amino acid acceptor D.
3.1 Endo M
Investigations began with Endo M. As mentioned previously, Endo M is a member of family GH 85 of the glycoside hydrolyases which cleaves both complex and high mannose N-glycans and which was first isolated from Mucor Hiemalis by Prof. Kenji Yamamoto (Kyoto) [12] and coworkers. Subsequent studies by the Yamamoto group revealed the ability of Endo M to catalyse transglycosylation reactions using full length N-glycans, albeit with low efficiency due to competing hydrolysis reactions [16]. Prof. Yamamoto later cloned and expressed Endo M in E. coli, and kindly donated samples of recombinant Endo M to us for use in a variety of glycosylation experiments using a wide range of structurally different donors.
3.1.1 Natural glycans
Endo M catalysed glycosylation of D using the N-glycan disaccharide donor 15 occurred in a very high yielding and irreversible fashion (95%, Table 1, entry 1). However, as the core disaccharide donor structure was extended, the reaction became less efficient and product hydrolysis became more prevalent. Of the two trisaccharide donors, glycosylation with the (1 → 6) linked donor 17 was the more efficient (maximum 89% yield, no product hydrolysis). The corresponding (1 → 3) linked donor 16 gave only a 65% maximum yield of product, which was in fact slowly hydrolysed by Endo M. This trend of decreased synthetic efficiency and increased product hydrolysis continued as the oxazoline structure was extended further. Thus the tetrasaccharide donor 18 gave a maximum 36% yield of a product which was itself hydrolysed, and the high mannose hexasaccharide donor 19 gave only a maximum 24% yield of a product which was even more rapidly hydrolysed. Finally the reaction using the truncated complex-type hexasaccharide donor 20 reached a rather modest maximal value of 22% after 70 min, after which time product yield again decreased due to competitive product hydrolysis.
Glycosylation catalysed by Endo M with natural N-glycan donors.
| Entry | Oxazoline donor | Max yield (%) | Time/min. | Product hydrolysis |
| 1 | 15 Disaccharide | 95 | 45 | No |
| 2 | 16 (1 → 3) Trisaccharide | 65 | 90 | Yes (slow) |
| 3 | 17 (1 → 6) Trisaccharide | 89 | 90 | No |
| 4 | 18 Tetrasaccharide | 36 | 180 | Yes |
| 5 | 19 High mannose hexasaccharide | 24 | 180 | Yes |
| 6 | 20 Complex hexasaccharide | 22 | 70 | Yes |
3.1.2 Non-natural glycans
Table 1 clearly shows that the utility of Endo M as a catalyst for the transfer of larger oligosaccharide structures is compromised by product hydrolysis. Attention therefore turned to the potential use of the gluco containing non-natural oxazolines (Fig. 3). The rationale for this study was the product oligosaccharides should not be cleaved by the endohexosaminidase since they contain a non-natural Glcβ(1 → 4)GlcNAc linkage adjacent to the cleavage site. However, the activated nature of the oxazoline could mean that these materials would still be substrates for the enzyme-catalysed glycosylation reaction itself; thus irreversible1 enzyme catalysed glycosylation, or glycoligation, may be achievable.
Glycosylation with the gluco disaccharide 21 only proceeded in very low 4% yield (Table 2, entry 1). However increasing the size of the oligosaccharide donor increased the synthetic efficiency of the process significantly and most notably none of the product oligosaccharides were observed to be hydrolytic substrates for Endo M. Thus glycosylation using the gluco (1 → 3) linked trisaccharide donor 22 proceeded in an excellent 88% yield in an irreversible process. In contrast to this promising result, use of the gluco (1 → 6) linked trisaccharide donor 23 only proceeded in a very low 4% yield, albeit in an irreversible process. This result indicated the importance of the (1 → 3) mannose linkage, which was presumably largely responsible for enhanced synthetic efficiency by binding to the Endo M active site. Finally use of the gluco tetrasaccharide 24 as a donor produced the desired pentasaccharide product in 70% yield in an irreversible fashion, a result which represented a considerable improvement on the efficiency of the reversible process using the corresponding manno donor 18 which only gave a maximum product yield of 36%. It was concluded that in all the cases assayed, the use of gluco containing oxazoline donors with Endo M resulted in irreversible glycosylation but that the efficiency of the reaction was crucially dependent on the oxazoline structure; this must contain the Man(1 → 3)Glc linkage in order for the donor to be processed by Endo M in a synthetically useful fashion.
Glycosylation catalysed by Endo M with non-natural gluco containing N-glycan donors.
| Entry | Oxazoline donor | Max Yield (%) | Time/min. | Product hydrolysis |
| 1 | 21 Gluco disaccharide | 4 | 180 | No |
| 2 | 22 (1 → 3) Gluco trisaccharide | 88 | 120 | No |
| 3 | 23 (1 → 6) Gluco trisaccharide | 4 | 180 | No |
| 4 | 24 Gluco tetrasaccharide | 70 | 60 | No |
3.2 Endo A
Attention then turned to the use of the other readily available family GH85 enzyme, Endo A. Endo A, which cleaves high mannose and hybrid N-glycans, was initially isolated from Arthrobacter protophormiae by Takegawa and coworkers [13]. Like Endo M, Endo A was found to be remarkable for its ability to catalyse transglycosylation reactions using full length Asn-linked N-glycans, albeit with low efficiency due to competing hydrolysis reactions [15]. During the course of these investigations, Wang and coworkers began publishing their studies on Endo A [19]. In light of these publications, the focus of our work became a systematic survey of the array of natural and non-natural donors available to us in order to build up a complete picture of structure–activity relationships.
3.2.1 Natural glycans
In the natural N-glycan series, the disaccharide donor 15 (Table 3, entry 1) produced the corresponding trisaccharide in quantitative yield after 30 min, though it was found that the product was slowly hydrolysed by Endo A if the reaction was left to run for much longer time periods. This result contrasted with the original findings of Shoda and coworkers [17], who reported that using the same donor 15 gave a product which did not undergo hydrolysis but which was produced in a much lower 44% yield. The study undertaken by us revealed that hydrolysis of the product did take place, but was only observed after reaction times of greater than 180 min. The use of the manno (1 → 3) trisaccharide donor 16 (Table 3, entry 2) also proceeded in excellent yield (96%), though again was reversible; the product was a substrate for slow Endo A catalysed hydrolysis, and the product yield decreased after reaching a maximum after 30 min. Glycosylation with the manno (1 → 6) trisaccharide donor 17 proceeded in a similar fashion, with a maximum yield of 76% after 20 min, and notably the rate of product hydrolysis was higher than in the case of the (1 → 3) linked donor 16. Glycosylation with the manno tetrasaccharide 18, a reaction reported by Wang prior to our study, was also efficient (maximum product yield of 88%), but also reversible. The high yield obtainable in this instance, together with the time course study, confirmed that the manno oxazoline 18 was a much better donor substrate for Endo A than it was for Endo M (vide supra), and in addition that reaction product was hydrolysed more rapidly by Endo M than it was by Endo A. However, it was found that, just as for Endo M, extension of the donor structure further led to marked decreases in the efficiency of the synthetic reaction. For example, the high mannose hexasaccharide 19 gave only a 15% maximum product yield. Moreover, this maximum was observed after only 12 min of reaction time indicating the extremely rapid rate of product hydrolysis effected by Endo A. Finally, use of the complex hexasaccharide donor 20 proceeded in only 11% yield, though in this last case no product hydrolysis was observed. The low yield for this reaction is perhaps not surprising since Endo A is not capable of cleaving complex N-glycans. Likewise, the lack of product hydrolysis for this more extended structure, which contrasts with the trends observed for the high mannose glycans and also for the corresponding reaction catalysed by Endo M, is also easily explicable in this instance.
Glycosylation catalysed by Endo A with natural N-glycan donors.
| Entry | Oxazoline donor | Max yield (%) | Time/min. | Product hydrolysis |
| 1 | 15 Disaccharide | 100 | 30 | Yes (slow) |
| 2 | 16 (1 → 3) Trisaccharide | 96 | 30 | Yes (slow) |
| 3 | 17 (1 → 6) Trisaccharide | 76 | 20 | Yes |
| 4 | 18 Tetrasaccharide | 88 | 30 | Yes |
| 5 | 19 High mannose hexasaccharide | 15 | 12 | Yes |
| 6 | 20 Complex hexasaccharide | 11 | 150 | No |
3.2.2 Non-natural glycans
Turning to the non-natural donors, use of the gluco disaccharide 21 with Endo A gave the corresponding product in 48% yield in an irreversible process (Table 4, entry 1). This result contradicted an earlier report from Wang and coworkers [19d] who had previously indicated that oxazoline 21 was not a substrate for Endo A catalysed glycosylation. In fact, the findings reported by us indicated that not only was 21 indeed a substrate for Endo A, but that Endo A was in fact more tolerant of a gluco unit in a disaccharide donor than Endo M (corresponding maximum 4% yield of product, Table 2, entry 1). Pertinently a subsequent reexamination of the reaction [19k] by Wang and coworkers revealed that Endo A did indeed catalyse glycosylation using this gluco oxazoline donor 21, although considerably larger quantities of enzyme were required to effect this reaction at a reasonable rate than they had employed in their first study. Moreover, another significant result from this later investigation was the observation that the use of large excesses of donor and quantities of enzyme could result in a controlled polymerisation process. It had previously been demonstrated that the 4-hydroxyl of glucose can in fact act as an acceptor for transglycosylations catalysed by both Endo A [13d] and Endo M [12g]. Therefore, there is no reason why in principle multiple additions cannot be effected by either of these enzymes. It is worthy of mention that mannose can also act as an acceptor for transglycosylations catalysed by either enzyme, and therefore the potential for double or even multiple glycosylation when using high mannose oxazoline donors cannot be completely excluded.
Glycosylation catalysed by Endo A with non-natural gluco containing N-glycan donors.
| Entry | Oxazoline Donor | Max yield (%) | Time/min. | Product hydrolysis |
| 1 | 21 Gluco disaccharide | 48 | 2000 | No |
| 2 | 22 (1 → 3) Gluco trisaccharide | 57 | 1500 | No |
| 3 | 23 (1 → 6) Gluco trisaccharide | 38 | 520 | No |
| 4 | 24 Gluco tetrasaccharide | 67 | 330 | Yes (very slow) |
Further investigations revealed that Endo A was indeed capable of processing all modified donors in which the gluco for manno substitution proximal to the N-acetyl glucosamine had been made (Table 4). Endo A catalysed glycosylation with the gluco (1 → 3) linked trisaccharide 22 proceeded in 57% yield, again in an irreversible process contrasting with the result obtained for the corresponding manno donor. The use of gluco (1 → 6) linked trisaccharide 23 with Endo A proceeded in a moderate 38% yield again in an irreversible fashion. Finally, use of the gluco tetrasaccharide 24 produced the desired pentasaccharide product in 67% yield, but in this final case extended time course studies revealed that Endo A was in fact capable of hydrolysing this gluco product. This result indicates that whilst the manno to gluco substitution does appear to oblate the hydrolytic capabilities of the enzymes for truncated glycan substrates, there is no guarantee that this substitution will completely negate product hydrolysis with more extended structures.
3.3 Endo H
The potential use of a variety of oxazoline donors in glycosylation reactions catalysed by Endo H was also investigated. Endo H [31], which cleaves high mannose and hybrid glycans, is a family GH18 enzyme, and therefore belongs to a different family of the glycoside hydrolases than Endo A and Endo M. Although Shoda et al. had reported [17] that Endo H was not capable of effecting glycosylation using the disaccharide donor 15, it was thought to be worthwhile investigating whether larger oxazolines could act as donor substrates. Indeed, the transglycosylation activity of Endo H had been previously demonstrated with full length N-glycans by Trimble et al. [32] who reported that glycerol may act as an acceptor, albeit in low yield and less efficiently than the corresponding reaction with Endo F.
Our studies, in fact, confirmed that that the Shoda observation was extendable to other oxazoline substrates, i.e. that Endo H was not capable of catalysing synthetic reactions using any of the oxazoline donors that were tested. There could be two reasonable explanations for this observation; either that direct oxazoline enzyme-catalysed hydrolysis is so rapid that no effective glycosylation of the GlcNAc acceptor can be seen, or alternatively that the oxazolines tested were simply not substrates for the enzyme. Since unfortunately the assay used in these experiments did not permit the concentration of the oxazoline donor to be followed accurately, no concrete conclusions can be drawn other than that the synthetic reactions did not work.
3.4 Variation of other reaction parameters
3.4.1 Effect of organic co-solvent
Previous reports into the use of Endo A [13d] and Endo M [33] for transglycosylation using Asn-linked donors detailed that increased synthetic efficiency could be achieved by the addition of an organic co-solvent to the aqueous reaction mixture. Indeed, the inclusion of an organic co-solvent has frequently been used to increase the synthetic efficiency of glycosidase-catalysed processes. Since the increased efficiency thereby observed is primarily due to a decrease in the rate of competitive hydrolytic processes, then the addition of quantities of organic co-solvent to the synthetic reactions catalysed by endohexosaminidases appeared to be a promising avenue for investigation.
The effect of the addition of an organic co-solvent on the synthetic efficiency of processes carried out using natural tetrasaccharide oxazoline donor 18 was investigated [18f]. Time course studies of synthetic reactions catalysed by Endo A and Endo M performed in the presence of varying quantities of DMSO as an organic co-solvent in phosphate buffer at pH 6.5 are shown in Fig. 4. For Endo A (Fig. 4A) the addition of 5% DMSO by volume to the reaction medium led to a significant improvement in the efficiency of the reaction. Quantitative conversion to product was observed after 30 min and no significant product hydrolysis occurred until after 180 min. Further experiments in which the DMSO content was increased to 10 and 15% led to further small decreases in the rate of product hydrolysis, but also to corresponding decreases in the rate of the synthetic reaction. In contrast for Endo M (Fig. 4B), an increase in the DMSO content actually led to a slight increase in the rate of the synthetic reaction, and correspondingly, the rate of product hydrolysis was also increased. Two other co-solvents were investigated for Endo A catalysed reactions. The use of either 10% acetone by volume or 10% DMF by volume was also found to have a beneficial effect; in both cases, the rate of the synthetic reaction was not greatly affected, but the rate of product hydrolysis was significantly diminished. It was concluded that the use of 10% acetone by volume probably represented the optimal reaction solvent for WT Endo A catalysed glycosylation reactions using oxazoline donors, whilst for Endo M the use of an organic co-solvent has no particularly beneficial effect.
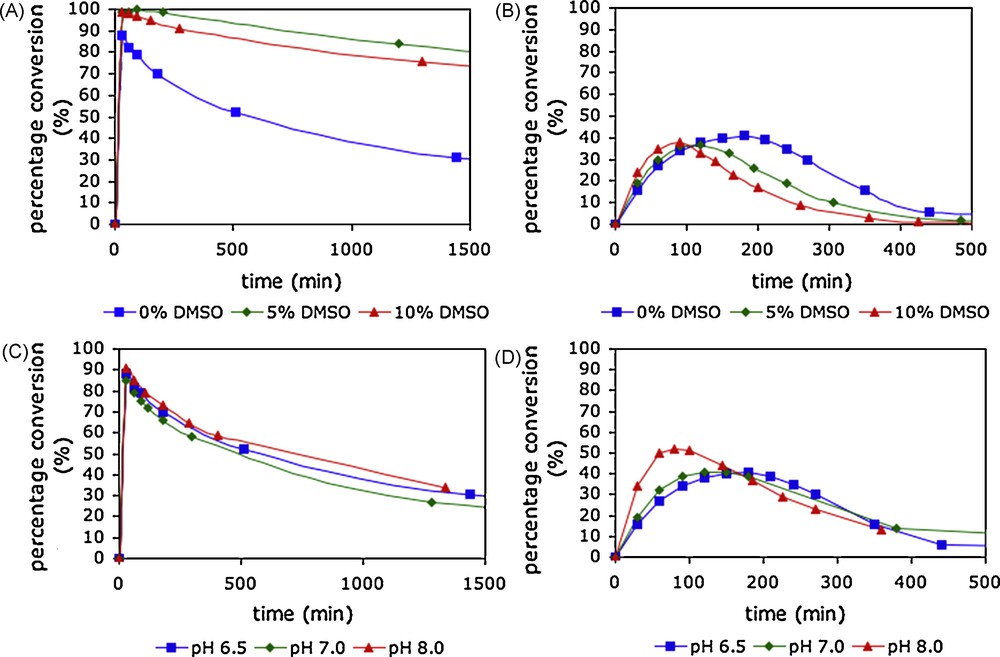
Time correlations of product yield for glycosylation of acceptor D by donor 18 with varying quantities of organic co-solvent and at different reaction pH; A) Endo A with varying organic co-solvent; B) Endo M with varying organic co-solvent; C) Endo A at varying reaction pH; D) Endo M at varying reaction pH.
3.4.2 Effect of pH
In several reports on the use of oxazolines as donors for endohexosaminidase-catalysed glycosylations, the reaction pH has been varied without specific comment. The pH/rate dependency for the hydrolytic activity of hexosaminidases is typically a bell-shaped curve centred around pH 5.0–8.0, the precise pH optimum depending on the enzyme [34]. The upper and lower limits of this bell-shaped curve generally correspond with the kinetic pKa values of the two catalytic acid residues in the enzyme active site. It is of note that family GH85 endohexosamimidases, such as Endo M and Endo A, possess only one key catalytic acid residue in the active site (E173 for Endo A, and E177 for Endo M). Published data for Endo M [12b] reported a pH rate maximum for hydrolytic activity in the range pH 6.0–6.5; at higher pH, the activity dropped markedly. In contrast the pH rate profile of Endo A was reported to have a very broad optimum, most active in the range pH 5.0–9.0, though the rate of hydrolysis was little reduced even at pH 11 [13a]. A brief study was undertaken [18f] to elucidate how synthetic efficiency varied with reaction pH for both of these enzymes using oxazolines as donors, the rationale being that moving to higher reaction pH may curtail the hydrolytic activity of the enzyme and allow greater synthetic efficiency to be achieved. Fig. 4 shows the pH dependency of glycosylation reactions catalysed by Endo A and Endo M. For Endo A (Fig. 4C) there was in fact no significant variation in product yield, or even in the time dependency of that yield, as the pH was increased from 6.5 to 8.0.2 This result reflects the broad pH optimum with effectively no upper limit reported for the hydrolytic activity of Endo A [13a]. However, Endo M (Fig. 4D) showed a more marked dependence on reaction pH. At pH 6.5, glycosylation of acceptor D with donor 18 gave a maximum product yield of 41% after 3 h. At higher reaction pH, the product was produced more rapidly, and the maximum obtainable yield was slightly increased. Thus, at pH 8.0, the maximum product yield was found to be 52% after 80 min, though after this time product hydrolysis led to a rapid decrease in yield. It can be concluded that whilst close control of reaction pH in the range 6.5–8.0 is not critical for obtaining the maximum synthetic yield for reactions catalysed by Endo A, working at higher pH does slightly increase the synthetic efficiency of reactions catalysed by Endo M. These results are broadly in line with the different pH/rate dependences and maxima previously described for these two enzymes.
3.5 Mutant enzymes
The above discussion highlights the continued problem that product hydrolysis represents. Attempted variation of reaction parameters had a limited effect, and did not represent a solution. Moreover, although the use of gluco containing oxazoline donors did, in most cases, prevent product hydrolysis, their use necessarily results in the production of non-natural glycans; a factor which may have important biological consequences in the context of glycoprotein therapeutics, for example, in eliciting undesired immune response. Consideration was therefore given to another potential way to circumvent the hydrolytic problem and the use of specifically mutated enzymes, or ‘glycosynthases’, as originally developed by Withers [35] and Planas [36], appeared to be an extremely attractive proposition. The term ‘glycosynthase’ was first applied by Withers to retaining glycosidases in which the nucleophilic catalytic acid residue in the enzyme active site had been replaced by site directed mutagenesis with a non-participating residue, for example, by alanine. The use of an activated glycosyl donor, such as a glycosyl fluoride, then allowed this mutant enzyme to promote a synthetic reaction, but the mutant enzyme was not capable of hydrolysing the product glycosidic linkage as the key nucleophilic residue was absent.
Since the Endo A plasmid was readily available, production of mutants of Endo A appeared to be an exciting prospect for curtailing the competitive hydrolytic reaction. However, the Withers/Planas glycosynthase approach cannot be applied by direct analogy to endohexosaminidases such as Endo A, as members of family GH85 catalyse hydrolysis via a neighbouring group participation mechanism in which the carbonyl oxygen of the 2-acetamide of the second GlcNAc residue is the actual nucleophile, rather than an enzyme-bound aspartate or glutamate. These enzymes therefore do not possess a nucleophilic residue at the active site to mutate out.
An alternative approach was therefore investigated in which the key catalytic acid residue of Endo A, glutamate 173, was replaced with an alternative. Previous mutational studies [13f] on Endo A had revealed that replacement of E173 with either glycine, aspartate, or glutamine had resulted in either extremely significant or complete loss of hydrolytic activity, whilst replacement with alanine produced a hydrolytically inactive mutant, the activity of which could be rescued by the addition of azide or formate. We reasoned that mutants of Endo A that lacked the general acid E173, although hydrolytically impaired, should still be able to process activated glycosyl donors, such as oxazolines. Consideration of the putative enzyme mechanism is informative (Scheme 5). In the first step of the hydrolytic mechanism, E173 acts as the general acid responsible for protonation of the outgoing β-glycosidic oxygen resulting in the formation of an intermediate oxazolinium ion (Scheme 5A). In the second step, E173 acts as a general base, de-protonating incoming water. Without a proton donor residue at position 173, all hydrolytic activity should be curtailed since glycosidic bonds cannot be broken without prior protonation of the anomeric oxygen. However, an oxazoline may still be able to enter into the mechanistic pathway and could still glycosylate an incoming alcohol (or be hydrolysed by incoming water) in the second step. Such a mutant enzyme would therefore be a glycosynthase; all hydrolytic activity is curtailed, but an activated oxazoline donor would still be processed.
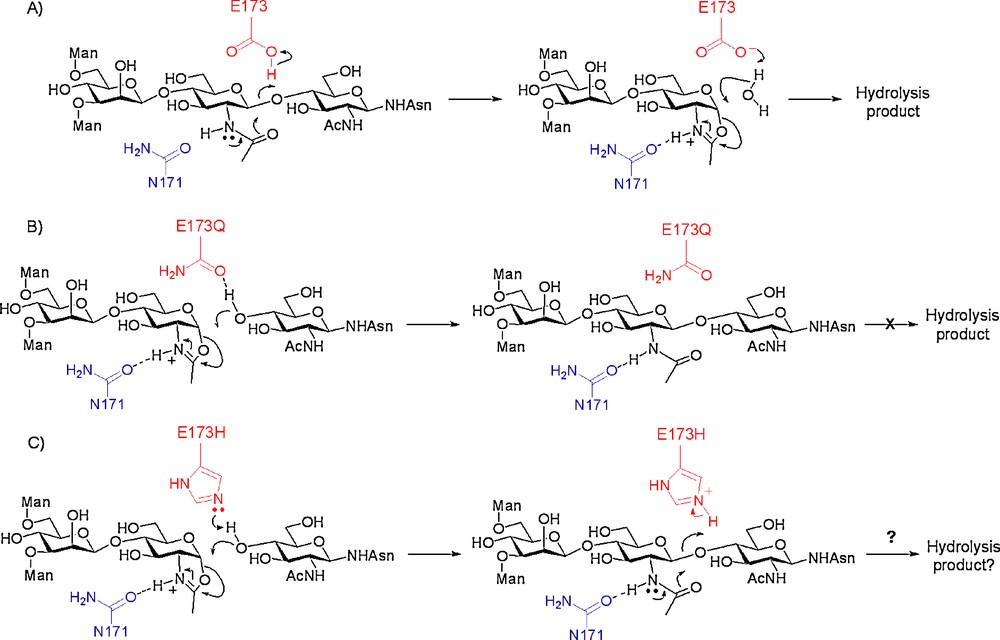
Catalytic mechanisms of Endo A; A) hydrolytic mechanism of WT Endo A; B) putative synthetic mechanism of mutant E173Q with an oxazoline donor; C) putative synthetic/hydrolytic mechanism of mutant E173H with an oxazoline donor.
Two mutants of Endo A were made in which E173 had been replaced by alternative amino acids; these were chosen as glutamine, a substitution which in fact had previously been made [13f] and which had resulted in total loss of hydrolytic activity, and histidine. Glutamine is a non-acidic residue and therefore should be incapable of promoting the hydrolytic reaction. The caveat was that it would also not be able to act as a general base and facilitate the synthetic reaction by aiding de-protonation of the incoming nucleophile, though it was thought possible that it could still act as a hydrogen bond acceptor and facilitate nucleophilic attack on the oxazoline (Scheme 5B). Alternatively, histidine could either act as a general acid, or as a general base, depending on its protonation state. The pKa of protonated histidine is ∼ 6.0 as compared to ∼ 4.1 for the side chain acid of glutamic acid; the hope was therefore that a histidine residue at position 173 would perhaps still act as a general base and facilitate the synthetic reaction, but that the higher pKa of its conjugate acid could mean a reduction in its ability to act as a general acid, with an accompanying reduction in the hydrolytic capability of the enzyme (Scheme 5C).
The ability of the E173Q and E173H mutants to catalyse glycosylation was assessed using the same assay as applied previously, and direct comparison with the wild-type (WT) enzyme undertaken [18d]. Time course studies of the reactions using the tetrasaccharide oxazoline donor 18 are shown in Fig. 5A. WT Endo A catalysed rapid product formation, the maximum obtainable yield being 88%, though after this time product hydrolysis was also rapid. In contrast, the E173H mutant catalysed a slightly slower synthetic reaction in which the essentially quantitative conversion to the product was achieved after ca. 2.5 h and after this time only extremely slow product hydrolysis was observed. Glycosylation catalysed by the E173Q mutant was considerably slower, and reaction times of about 20 h were required in order to reach the maximum yield of 82%. However, the E173Q enzyme, regardless of how long the reaction was left to run, did not hydrolyse the reaction product. These results indicated that that the two mutations had indeed suppressed the ability of the enzyme to catalyse product hydrolysis, but had not removed their ability to process oxazolines as activated donors. Although the E173H mutant did still retain some minor amount of hydrolytic activity, this was considerably reduced compared to that of WT Endo A, and replacement of glutamic acid 173 by glutamine had completely abolished product hydrolysis. Interestingly, the E173Q mutant had been previously reported not to possess transglycosylation activity when Asn-linked oligosaccharides were used as donors [13f], further underlining the utility of oxazolines as donors in these enzyme-catalysed glycosylations.

Time correlations of product yield for: A) glycosylation of acceptor D by donor 18 catalysed by WT Endo A and the E173H and E173Q mutants; B) glycosylation of dRNAse B with donor 18 catalysed by WT Endo A and the E173H mutant.
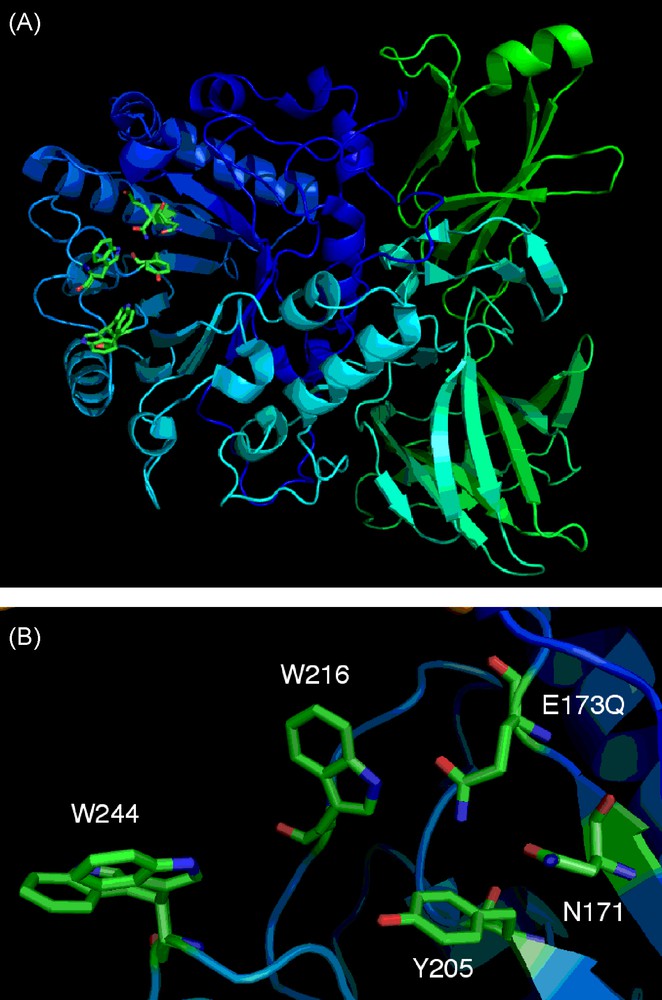
Having produced these two mutants, the Endo A E173Q mutant was successfully crystallised and its structure solved (Fig. 6) [37]. This was the first structure to be solved (two other family GH 85 structures have now also been solved WT Endo A [38a] and Endo D [38b]) for a family GH85 enzyme and deposited at the PDB.3 This structure, along with another structure solved for WT Endo A [38a] slightly later, provided confirmation of the identity and spatial arrangement of essential active site amino acids, such as residues E173Q, N171 and the proximity of hydrophobic residues Y205, W216, and W244 that had previously been identified as important for transglycosylation activity. These GH85 structures will also undoubtedly provide other key insights that may be used to optimise the synthetic activity of other Endo A mutants produced by future protein engineering.
Finally, it should be noted that Yamamoto and Wang undertook [19f] mutational studies on Endo M that were concurrent with our work on Endo A. Herein they identified an N175A mutant of Endo M in which asparagine 175, a conserved residue in the GH85 family, was replaced by alanine that displayed useful glycosynthase activity. Since this N175A mutant displayed only marginal hydrolytic activity, it was concluded that N175 plays a key role in promoting oxazolinium ion intermediate formation during the hydrolytic mechanism, though not by acting as the proton donor (identified for Endo M as E177). Wang has subsequently performed analogous mutations on Endo A [19l], which also produced a useful N171A glycosynthase mutant that has latterly been applied in glycoprotein remodelling.
3.6 Glycoprotein remodelling
The long-term motivation for undertaking these studies is the future development of robust procedures for the remodelling of proteins and mABs, both for therapeutic use and biochemical study. It will therefore not be lost on the reader that all the above studies have been performed on a model glycosyl amino acid acceptor. An important question is therefore whether the conclusions drawn from, and the advances made by such studies will be extendable to proteins as substrates.
Wang was the first to report the use of Endo A for the remodelling of a glycoprotein (RNase B) using oxazolines as donors [19e]. RNase B therefore served as an appropriate benchmark for examination of the utility of the remodelling procedure. Given the Wang precedent, our attention turned to the potential application of our mutant enzymes for protein glycosylation. Commercially available bovine RNase B, containing a heterogeneous mixture of high mannose glycoforms, was first enzymatically trimmed back by treatment with Endo H to produce dRNase B, a single homogeneous protein glycoform bearing a GlcNAc residue at the sole N-linked glycosylation site (N34). dRNase B was then used as a substrate for glycosylation with tetrasaccharide oxazoline 18 using both WT Endo A and the E173H mutant (Scheme 6). In both cases [18d], the enzyme was able to effect production of a single glycoform product, Man3GlcNAc2RNase B, which was readily characterised by mass spectrometry. Time course studies of these reactions (Fig. 5B) revealed that both enzymes efficiently catalysed the formation of the product Man3GlcNAc2 glycoform of RNase B. The maximum yields obtainable were 72% using WT Endo A and 84% using the E173H mutant. This time course study revealed that WT Endo A effected transglycosylation of dRNase B more rapidly than the E173H mutant. However, WT Endo A also catalysed hydrolysis of the product glycoprotein, Man3GlcNAc2RNase B, whereas the E173H mutant did not. Therefore, after a certain time period, the E173H mutant became the more efficient catalyst and ultimately allowed formation of the glycoprotein product in a higher yield.
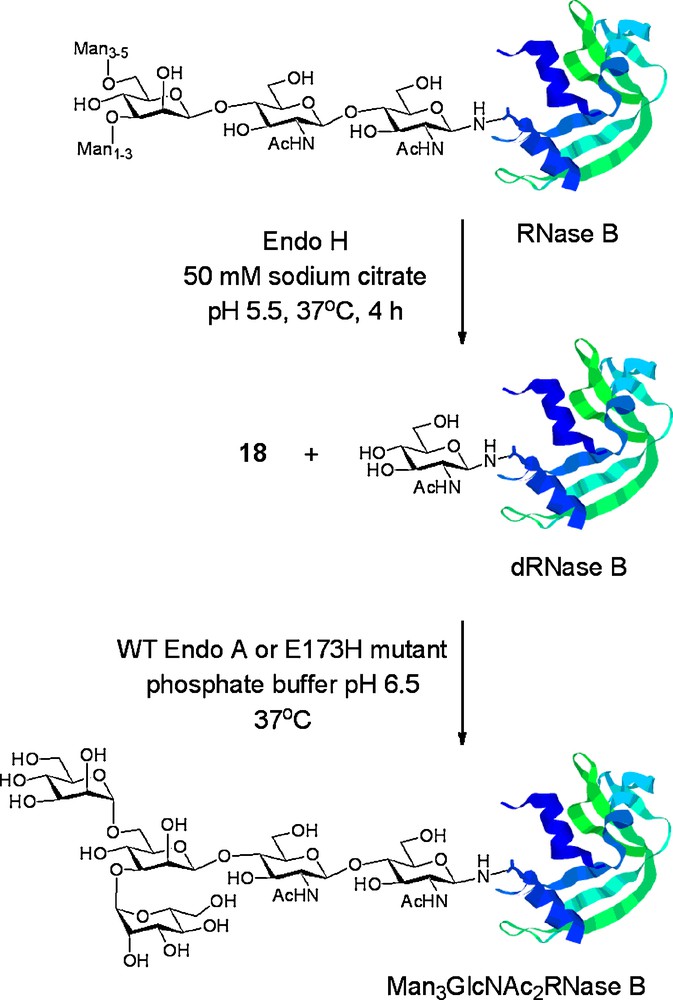
Enzymatic remodelling of a mixture of RNase B glycoforms to a single Man3GlcNAc2 glycoform using WT Endo A and the E173H mutant.
4 Conclusions and future perspectives
Members of family 85 of the glycoside hydrolases, such as Endo A and Endo M, show a remarkable ability to transfer oligosaccharides en bloc to glycoconjugates bearing GlcNAc residues. However, the utility of conventional transglycosylation using full-length Asn-linked N-glycans is quite limited as yields are poor even when large excess quantities of valuable glycans are employed. The use of oxazolines as activated donors can overcome this low efficiency, and shows exceptional promise as a means of facilitating access to specifically glycosylated glycoconjugates bearing defined homogenous N-glycan structures. A wide variety of N-glycan oxazoline structures may be readily accessed by chemical synthesis and, in general, these family GH85 enzymes are able to transfer these materials to acceptors bearing GlcNAc residues. However, with more extended glycan structures competitive product hydrolysis limits achievable synthetic efficiency, even using oxazolines as donors. Undesirable product hydrolysis may, however, be counteracted, either by use of structurally modified N-glycan oxazolines to produce non-natural products that the enzyme is not able to cleave, or by the production of mutant enzymes which are themselves not capable of product hydrolysis but which still process oxazolines as donors. Thus, this methodology may be employed to access a range of defined homogenous glycoproteins, such as RNase B, in a considerably more efficient fashion than can be achieved by alternative means, for example, by total synthesis.
Despite the important advances achieved so far, several issues need to be addressed if this type of process is to be widely applicable to a range of complex glycoconjugates, such as therapeutic glycoproteins and mAbs. One major limitation is that of direct enzyme-catalysed oxazoline hydrolysis. This undesired reaction means that excesses of oxazoline donors are always required, and that reactions become considerably less efficient when proteins or other macromolecules are employed as the acceptors. Other not-inconsiderable challenges also remain for the future. For example, methodology will need to be developed which will allow the efficient attachment of different N-glycans to different glycosylation sites of a protein simultaneously [16f]. Furthermore, control of O-linked glycosylation is also an issue for future investigation. Such challenging objectives will continue to test the ingenuity of synthetic chemists for the foreseeable future. However, one thing is certain: the therapeutic and commercial importance of glycoproteins and mAbs will spur on even more intensive research efforts in this field.
Acknowledgements
I sincerely thank the members of my group, Dr Thomas Rising, Thomas Parsons and Dr Christoph Heidecke who performed all of the synthetic work detailed in this mini-review. I also thank my collaborators for their invaluable assistance: Dr James Moir, Dr Zhenlain Ling and Dr Ed Taylor (University of York) for production of WT Endo A and the E173Q and E173H mutants, and for solving the E173Q Endo A crystal structure; Professor K. Yamamoto (University of Kyoto) for kindly providing the Endo M enzyme; Professor K. Takegawa (University of Kagawa) for kindly providing the Endo A plasmid. I also gratefully acknowledge financial support from the BBSRC and the EPSRC.
1 The terms ‘reversible’ and ‘irreversible’ are only applied, for want of better wording, in a loose sense and apply only with respect to the glycosyl acceptor. This is because enzymatic product hydrolysis, whilst reforming the GlcNAcAsn glycosyl acceptor D, does not result in reformation of the oxazoline donor.
2 It was unfortunately not possible to extend the study to include reactions performed above pH 8.0 as decomposition of the acceptor D occurred at these higher pH values. Decomposition of D also meant that quantitative analysis of experiments performed at pH 8.0 for reaction times of greater than 7 hours was not always possible.
3 PDB accession number 2vtf. Deposition date: 14th May 2008.