

1 Introduction
Les apatites constituant une grande famille de composés inorganiques ont été intensivement étudiées ces dernières décennies. Cela est attribué à leurs diverses applications et à la faculté de leur structure à générer de nouveaux matériaux grâce aux diverses substitutions qu’elle peut accepter [1–4]. Ainsi, elles sont utilisées comme biomatériaux [5,6], catalyseurs [7,8] et matériaux pour laser [9,10]. De plus, la littérature rapporte continuellement de nouveaux matériaux dont les applications restent à découvrir.
Les apatites sont représentées par la formule générale M10(XO4)6Y2, où M est un cation bivalent (Sr2+, Ca2+, Pb2+, Cd2+, etc.), XO4 est un groupement anionique trivalent (PO43−, VO43−, AsO43−, etc.) et Y un monoanion (F−, Cl−, OH−, etc.). Elles cristallisent principalement dans le système hexagonal avec le groupe d’espace P63/m [11–13]. Un arrangement quasi-compact des groupements XO43− constituant le squelette de cette structure présente deux types de tunnels. Le premier, de symétrie locale C3, est occupé par quatre cations, notés M(1), qui sont entourés par neuf atomes d’oxygène. Le second tunnel présentant la symétrie Cs est plus large. Il est occupé par les six cations restants, notés M(2). Ces cations qui sont entourés par six atomes d’oxygène et un atome Y forment deux triangles équilatéraux alternés à la cote ¼ et ¾, centrés sur l’axe sénaire hélicoïdal.
En raison de la flexibilité de cette structure, les apatites peuvent s’accommoder de nombreuses substitutions, cationiques, anioniques ou les deux à la fois, conduisant à la formation de solutions solides totales ou partielles. Ainsi, la substitution d’un cation divalent par une terre rare trivalente (Ln3+) et le groupement XO43− trivalent par le groupement silicate tétravalent, SiO44−, conduit à la formation de composés, appelés britholites [14,15]. Ces composés ont suscité un grand intérêt parmi les chercheurs en raison de leurs applications potentielles comme conducteurs ioniques [16,17] ou matrice pour le confinement des sous-produits de l’industrie nucléaire tels que les actinides mineurs ou les produits de fission à vie longue [18–24], notamment sur la base de bonnes résistances à l’altération [25–26] et à l’irradiation [27–29]. De plus, ces matériaux sont largement étudiés pour leurs propriétés de luminescence [30,31].
Dans ce travail, nous nous sommes intéressés à la synthèse de fluorbritholites strontiques dopées au néodyme. Après avoir caractérisé les produits obtenus par diffraction des rayons X, spectroscopies de diffusion Raman et d’absorption infrarouge, nous avons procédé à l’étude de leurs propriétés optiques.
2 Protocole expérimental
2.1 Synthèse
Des fluorbritholites de formule générale Sr10xNdx(PO4)6-x(SiO4)xF2 avec 0 ≤ x ≤ 6, ont été préparées par réaction à l’état solide à partir de SrCO3 (Cerac, 99,99 %), SrF2 (Prolabo, ≥ 99,5 %), Nd2O3 (Acros, 99,9 %), SiO2 (alpha, ≥ 99,5 %), (NH4)2HPO4 (Prolabo, ≥ 99,0 %) et Sr2P2O7.
2.1.1 Préparation de Sr2P2O7
Le diphosphate de strontium a été préparé à partir de (NH4)2HPO4 et SrCO3 selon la réaction suivante :
| 2SrCO3 + 2(NH4)2HPO4 → Sr2P2O7 + 4NH3 (↑) + 2CO2 (↑) + 3H2O (↑) | (1) |
Après avoir été broyés et homogénéisés dans un mortier en agate, les réactifs ont été mis en forme par pressage uniaxial à froid afin d’améliorer le contact entre particules et favoriser ainsi la réaction. Ensuite, les pastilles ont été calcinées à 1000 °C pendant 24 heures avec une vitesse de montée en température de 10 °C par minute.
2.1.2 Préparation des fluorbritholites
Les réactifs ont été pris dans des proportions stœchiométriques selon l’équation suivante :
| SrF2 + 3SrCO3 + (6-x)/2Sr2P2O7 + x/2Nd2O3 + xSiO2 → Sr10-xNdx(PO4)6-x(SiO4)xF2 + 3CO2 (↑) | (2) |
avec 0 ≤ x ≤ 6.
Après avoir été intimement mélangés dans un mortier en agate, les réactifs ont été, comme lors de la préparation de Sr2P2O7, pressés à froid sous forme de pastille de 30 mm de diamètre. Ils ont ensuite subi deux traitements thermiques sous atmosphère dynamique d’argon. Le premier traitement a été réalisé à 900 °C pendant 12 heures dans le but d’évacuer les produits de décomposition, alors que le deuxième a eu lieu à des températures comprises entre 1200 et 1400 °C pendant des durées variant entre six et 12 heures, selon la teneur de l’échantillon en silicate. Pour tous les échantillons, ce dernier traitement a été appliqué à trois reprises, entrecoupées par une étape de broyage/homogénéisation et mise en forme. La vitesse de montée en température est de 10 °C par minute.
2.2 Techniques de caractérisation
L’identification des phases a été réalisée par diffraction des rayons X (DRX) au moyen d’un diffractomètre de marque PANalytical X’Pert Pro, utilisant la radiation Kα du Cobalt (λ = 1,789 Å). Le domaine angulaire entre 10 et 60°C en 2θ a été balayé par pas de 0,01°C et un temps de comptage de 18 s par pas. L’identification des phases cristallines a été réalisée par comparaison des diffractogrammes obtenus avec les fichiers Joint Commitee Powder Diffraction Standard (JCPDS) en utilisant le logiciel X’Pert HighScore Plus. Les paramètres de maille ont été déterminés par la méthode des moindres carrés.
Les spectres de diffusion Raman ont été enregistrés à l’aide d’un spectromètre Micro-Raman Renishaw RM 1000, muni d’une source laser YAG : Nd3+ doublé en fréquence, émettant à 532 nm, et équipé d’un multi-détecteur de type CCD.
Les spectres d’absorption infrarouge (IR) ont été acquis dans le domaine 1350–450 cm−1, au moyen d’un spectromètre à transformée de Fourier de marque Perkin Elmer GX 2000, en utilisant des pastilles préparées à partir d’un mélange homogène de 1 mg de produit et 300 mg de KBr.
Les mesures d’absorption optique ont été effectuées dans les domaines proche-IR et visible à l’aide d’un spectrophotomètre doté d’une sphère intégratrice de type USB2000-VIS-NIR Ocean Optics.
Les spectres de luminescence ont été enregistrés dans le domaine spectral 1000–1400 nm au moyen d’un spectromètre Spectra-Physics 2018 équipé d’une source Laser Ar-Kr émettant à 514,5 nm. L’excitation émise est dispersée par un monochromateur Jobin-Yvon HRD1 ; la détection a été effectuée à l’aide d’un photomultiplicateur Hamamatsu HV 1250.
3 Résultats et discussion
3.1 Diffraction des rayons X
Les diagrammes DRX des échantillons avec 0 ≤ x ≤ 6 sont présentés sur la Fig. 1. Ils ont tous été identifiés par référence à la fiche JCPDS no 00-50-1744, correspondant à la fluorapatite strontique de symétrie hexagonale et de groupe d’espace P63/m. Pour x ≤ 2,5, les raies de diffraction de chaque composition appartiennent à la phase apatitique et aucune raie supplémentaire, susceptible d’appartenir à l’un des réactifs de départ ou à une quelconque phase secondaire, n’a été mise en évidence (Fig. 1a). Pour x ≥ 3, les diffractogrammes renferment, en plus de celles de la phase apatitique, des raies supplémentaires dont les intensités augmentent avec x (Fig. 1b). Elles correspondent aux phases Sr2SiO4 et Nd2SiO5 dont les fiches JCPDS sont respectivement no 01-076-1630 et no 00-040-0284. Ces phases seraient formées respectivement selon les réactions suivantes :
| SiO2 + 2SrCO3 → Sr2SiO4 + 2CO2 (↑) | (3) |
| SiO2 + Nd2O3 → Nd2SiO5 | (4) |
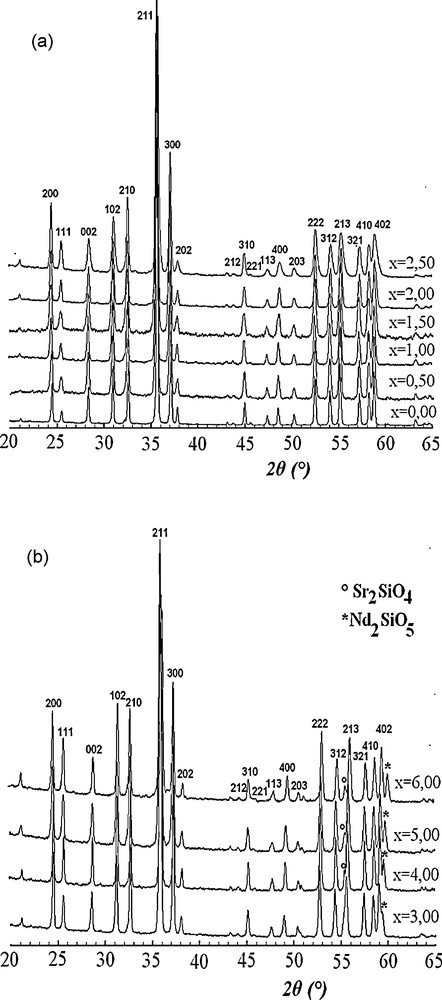
Diagrammes DRX des échantillons de formule Sr10-xNdx(PO4)6-x(SiO4)xF2 (a) 0 ≤ x≤ 2,5 ; (b) x ≥ 3.
En comparant les positions relatives des raies DRX de chaque échantillon substitué aux positions des raies de la fluorapatite pure, on constate qu’elles se déplacent progressivement du coté des angles élevés au fur et à mesure que l’apatite s’enrichit en substituants. Ce déplacement, consécutif à la contraction de la maille, peut servir de preuve à l’incorporation des ions Nd3+ et SiO44− dans la structure apatitique. Les paramètres de maille, a et c, et le volume correspondant sont reportés dans le Tableau 1, tandis que leurs variations en fonction de x sont respectivement illustrées aux Fig. 2 et 3. Comme le montrent ces figures, les paramètres a et c ainsi que le volume de la maille diminuent linéairement et progressivement de part et d’autre de la valeur x = 3.
Paramètres cristallographiques des compositions étudiées.
| Compositions | a (Å) | c (Å) | V (Å3) |
| Sr10(PO4)6F2 | 9,713(3) | 7,287(7) | 595,369(2) |
| Sr9,5Nd0,5(PO4)5,5(SiO4)0,5F2 | 9,710(3) | 7,277(5) | 594,184(3) |
| Sr9Nd(PO4)5(SiO4)F2 | 9,707(4) | 7,269(3) | 593,164(4) |
| Sr8,5Nd1,5(PO4)4,5(SiO4)1,5F2 | 9,704(2) | 7,265(4) | 592,472(2) |
| Sr8Nd2(PO4)4(SiO4)2F2 | 9,702(4) | 7,259(2) | 591,738(2) |
| Sr7,5Nd2,5(PO4)3,5(SiO4)2,5F2 | 9,699(1) | 7,256(3) | 591,291(3) |
| Sr7Nd3(PO4)3(SiO4)3F2 | 9,697(2) | 7,251(5) | 590,477(3) |
| Sr6Nd4(PO4)2(SiO4)4 F2 | 9,669(5) | 7,226(3) | 585,048(3) |
| Sr5Nd5(PO4)(SiO4)5F2 | 9,643(2) | 7,203(4) | 580,053(4) |
| Sr4Nd6(SiO4)6F2 | 9,619(2) | 7,178(6) | 575,166(3) |
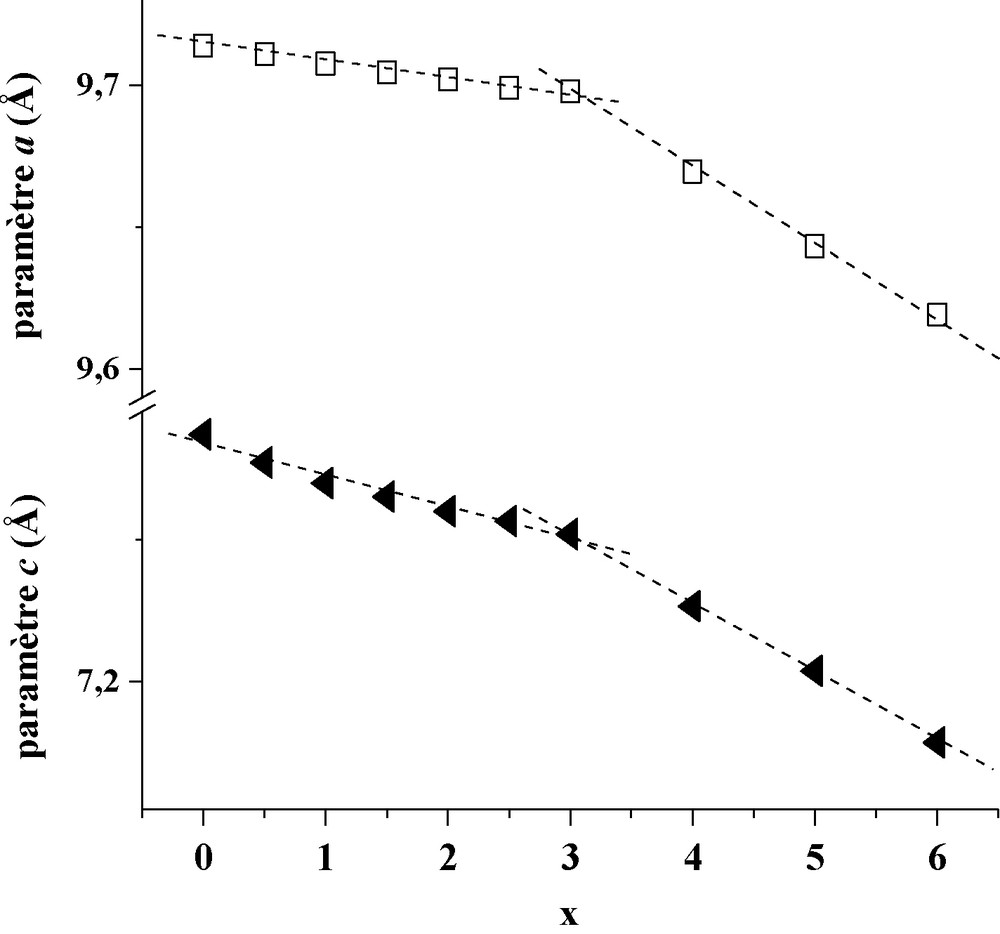
Variation des paramètres cristallographiques a et c en fonction de x.

Variation du volume V de la maille en fonction de x.
Pour 0 ≤ x ≤ 2,5, L’évolution linéaire des courbes obéit à la loi de Végard selon les équations suivantes :
| a = (9,713 – 0,006x) Å σ = 2,8 × 10−4 Å | (5) |
| c = (7,284 – 0,012x) Å σ = 4 × 10−4 Å | (6) |
| V = (595,139 – 1,654x) Å3 σ = 2,6 × 10−4 Å3 | (7) |
Dans ce domaine de composition, Δa/a et Δc/c ont comme valeurs respectives 0,09 et 0,27 %. On constate que la variation selon l’axe c est plus importante que selon l’axe a.
Pour x ≥ 3, bien qu’il y ait formation de phases secondaires (Sr2SiO4 et Nd2SiO5), l’évolution linéaire des paramètres cristallographiques en fonction de x indique que la solution solide continue à se former, mais une partie seulement des quantités de substituants utilisées est incorporée dans l’apatite.
La diminution des paramètres cristallographiques avec x peut être reliée à la taille des ions en substitution. Cependant, si Sr2+ (coordination 9 : rayon ionique = 1,31 Å) a été remplacé par un ion plus petit Nd3+ (coord. 9 : r.i. = 1,16 Å) [32], PO43− (dliaison P–O = 1,51 Å) quant à lui, a été substitué par un ion plus volumineux, SiO44− (dliaison Si–O = 1,62 Å).
Boughzala et al., préparant des britholites strontiques au lanthane, ont observé une évolution inverse des paramètres de maille, lorsque les quantités incorporées de La3+ et SiO44− augmentent : a croît et c diminue [33]. La solution solide formée est continue entre les compositions limites Sr10(PO4)6F2 et Sr4La6(SiO4)6F2. Par ailleurs, l’affinement structural de ces phases par la méthode de Rietveld [34] a montré, en accord avec des études antérieures [35–37], que les ions La3+ sont préférentiellement localisés dans les sites M(2) de la structure apatitique. Toutefois, cette tendance tend à s’atténuer avec l’augmentation de x. Dans la structure de l’apatite, l’axe a est plus influencé que l’axe c par les tailles respectives des sites M(2) et des tétraèdres XO4, alors que l’axe c est plutôt influencé par la taille des sites M(1).
L’évolution inverse du paramètre a dans les solutions solides au lanthane (a augmente) et dans leurs homologues au néodyme (a diminue) serait liée à la différence des tailles des deux cations qui sont respectivement incorporés dans les deux séries : La3+ (coord. 9 : r.i. = 1,21 Å) étant plus petit que Sr2+ (coord. 9 : r.i. = 1,31 Å), mais plus gros que Nd3+ (coord. 9 : r.i. = 1,16 Å). Ainsi, dans les solutions solides au lanthane, l’augmentation de a peut être expliquée par l’effet prépondérant sur l’axe a de la taille du groupement silicate devant celle de l’ion La3+, alors que dans la série au néodyme, c’est l’effet de la taille de l’ion Nd3+ qui devient prépondérant devant celui de la taille de SiO44−.
Quant à l’existence des deux domaines pour la solution solide de part et d’autre d’une valeur de x voisine de 3 environ, elle pourrait être due à une distribution différente des ions Nd3+ entre les deux sites cationiques M(1) et M(2). La détermination des taux d’occupation de ces sites par les ions Nd3+ pourrait contribuer à une meilleure compréhension de l’évolution des paramètres de maille en fonction de x et expliquer la discontinuité de la solution solide.
En raison de la formation de phases secondaires pour les compositions dont la teneur en substituants est supérieure à 2,5, nous nous sommes limités, dans la suite de ce travail, à l’étude des compostions pour lesquelles x est inférieur à cette valeur.
3.2 Spectroscopie de diffusion Raman
Les spectres acquis à la température ambiante, dans le domaine 350–1200 cm−1, sont représentés à la Fig. 4. Les attributions des bandes de vibration des groupements PO4 et SiO4 ont été effectuées par comparaison avec des spectres de phases apatitiques de compositions similaires, rapportés dans la littérature [38,39]. Le Tableau 2 regroupe, pour les différentes compostions, les positions et les attributions des différentes bandes.
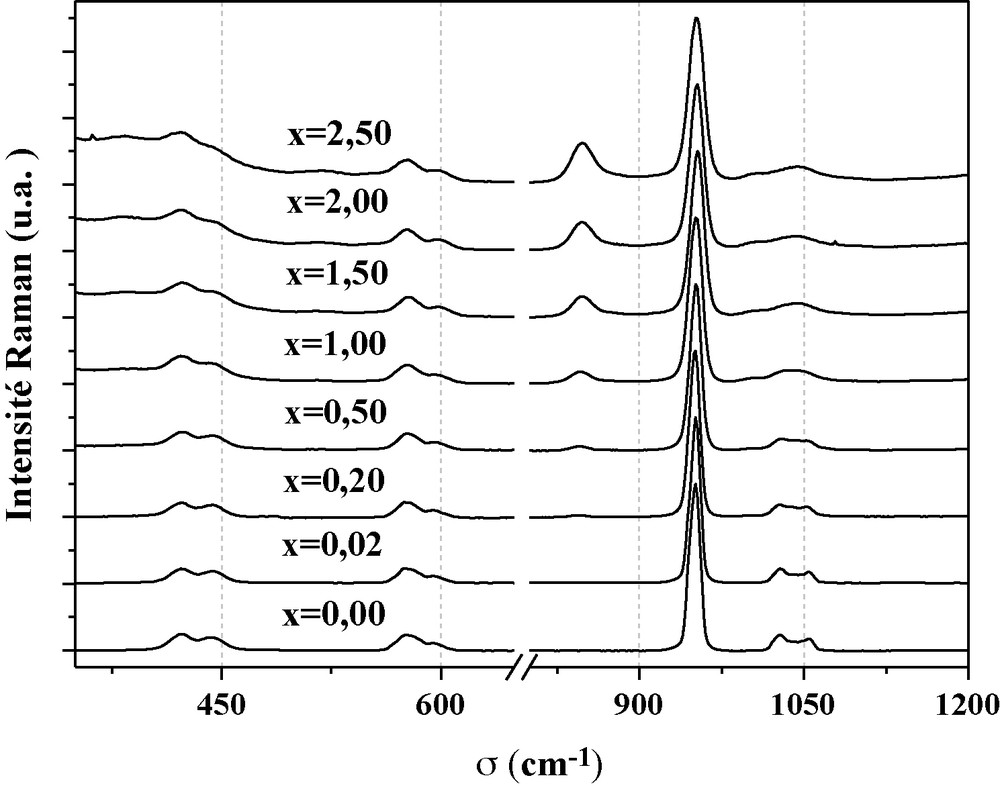
Spectres de diffusion Raman des britholites de formule Sr10-xNdx(PO4)6-x(SiO4)xF2 avec 0 ≤ x≤ 2,5.
Attribution et positions des bandes de diffusion Raman.
| Compositions | PO43− | SiO44− | ||||||
| (cm−1) | ν1 | ν2 | ν3 | ν4 | ν1 | ν2 | ν3 | ν4 |
| Sr10(PO4)6F2 | 952 | 442 421 | 1054 1028 | 595 576 | – | – | – | – |
| Sr9,98Nd0,02(PO4)5,98(SiO4)0,02F2 | 952 | 442 422 | 1054 1028 | 595 576 | – | – | – | – |
| Sr9,8Nd0,2(PO4)5,8(SiO4)0,2F2 | 952 | 443 422 | 1053 1028 | 595 576 | 845 | – | – | – |
| Sr9,5Nd0,5(PO4)5,5(SiO4)0,5F2 | 952 | 443 421 | 1053 1029 | 595 576 | 845 | – | – | – |
| Sr9Nd(PO4)5(SiO4)F2 | 952 | 442 421 | 1043 1001 1038 | 595 576 | 846 | 385 | 868 | – |
| Sr8,5Nd1,5(PO4)4,5(SiO4)1,5F2 | 952 | 443 423 | 1042 1005 | 598 578 | 848 | 385 | 863 | 517 |
| Sr8Nd2(PO4)4(SiO4)2F2 | 952 | 446 – | 1042 1008 | 597 576 | 848 | 385 | 865 | 517 |
| Sr7,5Nd2,5(PO4)3,5(SiO4)2,5F2 | 952 | 445 – | 1045 1008 | 598 576 | 848 | 384 | – | 520 |
Pour l’échantillon non substitué, les bandes associées aux groupements PO4 telles que celles observées à 952 cm−1 et entre 1053–1029 cm−1 sont respectivement attribuées aux modes de vibration symétrique, ν1 et antisymétrique ν3, alors que les bandes situées entre 421–442 cm−1 et 595–576 cm−1 correspondent respectivement aux modes de déformation symétrique ν2 et antisymétrique, ν4. En plus de ces bandes, les spectres des échantillons substitués présentent des bandes supplémentaires associées aux groupements SiO4. Elles sont respectivement situées à 845 cm−1 (ν1), 385 cm−1 (ν2), 865 cm−1 (ν3) et 517 cm−1 (ν4). Les intensités relatives de ces absorptions augmentent avec l’augmentation de x, confirmant ainsi l’incorporation des groupements silicate dans la structure apatitique.
Afin d’évaluer l’influence de l’incorporation de la paire d’ions, Nd3+ et SiO44−, sur la structure de l’apatite, nous avons suivi, en fonction de x, l’évolution de la largeur à mi-hauteur de la bande la plus intense, ν1, relative au groupement phosphate. Selon Weber et al. [40], la largeur effective est définie par la relation :
| (8) |
La Fig. 5 illustre l’évolution de la largeur effective Δλeff de cette bande en fonction de x. Comme le montre cette figure, Δλeff croît avec l’augmentation de la quantité de Nd3+ incorporée ; elle varie de 12,0 ± 0,1 cm−1 à 19,8 ± 0,1 cm−1 lorsque x varie respectivement de 0 à 2,5. Cet élargissement serait dû au fait que chaque mode de vibration ne correspond plus à une seule fréquence mais plutôt à une distribution de fréquences à cause du désordre de la structure apatitique. Par ailleurs, en comparaison avec d’autres structures apatitiques [41], la valeur Δλeff = 12 cm−1 correspondant à la composition phosphatée semble importante. Elle pourrait être due à une distorsion des tétraèdres PO4 ou encore à l’existence de lacunes cationiques. Dans ce cas, le couplage entre le groupement PO4 et ces défauts fait intervenir des modes de vibration locaux en plus des modes de vibration propres, contribuant ainsi à l’élargissement de cette bande. En outre, les impuretés que peuvent renfermer les réactifs utilisés peuvent introduire un certain désordre.

Évolution de la largeur effective Δνeff de la bande de diffusion Raman ν1 du groupement PO4 en fonction de x.
Avec l’incorporation de Nd3+ et SiO44−, il y a accroissement du désordre de la structure, et aux modes de vibrations des groupements PO4 évoqués précédemment, se superposent de nouveaux modes, localisés, dus au couplage des liaisons P–O avec leur environnement. De plus, la distribution des ions Nd3+ entre les deux sites cristallographiques M(1) et M(2) doit perturber profondément les modes normaux de vibration et contribuer alors à l’étalement de la largeur des bandes de diffusion.
3.3 Spectroscopie d’absorption infrarouge
Les spectres d’absorption infrarouge sont présentés sur la Fig. 6, et les valeurs des positions relatives de leurs bandes d’absorption sont consignées dans le Tableau 3. Leur attribution a été réalisée en se référant à la littérature [42,43]. Le spectre de l’échantillon non substitué montre les bandes caractéristiques de la fluorapatite. En plus des bandes relatives aux modes de vibrations des groupements PO4, les spectres des échantillons substitués présentent des absorptions associées aux groupements SiO4 situées à 839–870 (ν1), 997 (ν3) et 508–548 (ν4) cm−1. Leurs intensités augmentent tandis que celles des bandes relatives aux groupements phosphate diminuent au fur et à mesure que la quantité de silicate utilisée augmente. Il est à signaler que les modes ν1 et ν2 des groupements PO4 et SiO4 sont non actives en absorption IR. Cependant, en raison des distorsions de ces groupements, les modes ν1 et ν2 pour PO4, et le mode ν1 pour SiO4 deviennent observables. Comme pour les bandes de diffusion Raman, on note l’élargissement des bandes d’absorption IR qui serait dû à l’augmentation du désordre dans la structure apatitique, évoqué précédemment.
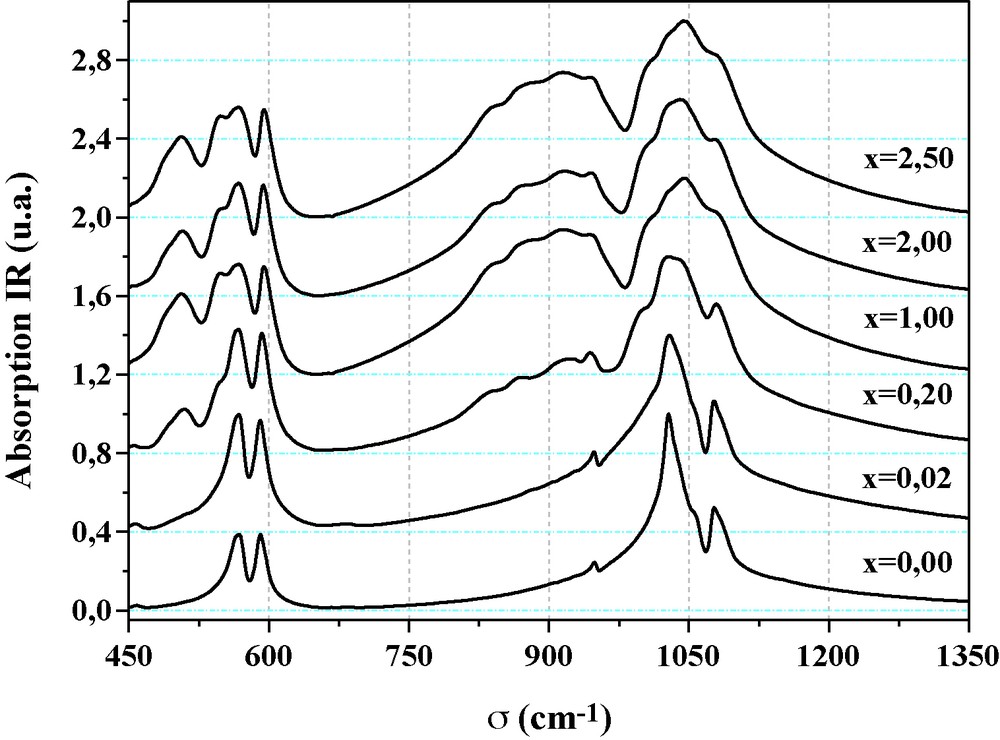
Spectres IR des britholites de formule Sr10-xNdx(PO4)6-x(SiO4)xF2 avec 0 ≤ x≤ 2,5.
Attribution et positions des bandes d’absorption infrarouge.
| Compositions | PO43− | SiO44− | ||||||
| (cm−1) | ν1 | ν2 | ν3 | ν4 | ν1 | ν2 | ν3 | ν4 |
| Sr10(PO4)6F2 | 948 | 458 – | 1028 1078 | 590 566 | – | – | – | – |
| Sr9,98Nd0,02(PO4)5,98(SiO4)0,02F2 | 947 | 457 | 1031 1079 | 590 567 | – | – | – | 506 – |
| Sr9,8Nd0,2(PO4)5,8(SiO4)0,2F2 | 945 | 455 | 1034 1080 | 592 567 | 837 869 | – | 997 | 508 543 |
| Sr9Nd(PO4)5(SiO4)F2 | 945 | – | 1035 1080 | 593 567 | 839 869 | – | 1003 | 507 548 |
| Sr8Nd2(PO4)4(SiO4)2F2 | 945 | 453 | 1039 1080 | 593 567 | 839 870 | – | 1003 | 508 548 |
| Sr7,5Nd2,5(PO4)3,5(SiO4)2,5F2 | 945 | – | 1043 1082 | 593 567 | 840 871 | – | 1005 | 508 548 |
3.4 Absorption optique
Dans le but de recueillir des informations complémentaires sur les changements structuraux intervenant dans la matrice étudiée, des mesures d’absorption optique ont été effectuées. Pour les échantillons substitués avec 0 < x ≤ 2,5, les spectres ont été acquis, dans le domaine 400–1000 nm, à la température ambiante en utilisant comme référence une poudre non dopée, de telle sorte que les seules raies d’absorption observées sont celles correspondant à l’ion Nd3+, et plus particulièrement aux transitions internes de la configuration 4f 3.
Sur la Fig. 7 est présenté le spectre d’absorption de l’échantillon pour x = 0,2, avec les attributions des différentes bandes ; l’état fondamental étant le niveau 4I9/2. La bande relative à la transition 4I9/2 → 4F5/2 + 2H9/2, centrée approximativement autour de 807 nm, est la plus intense. Elle présente une largeur à mi-hauteur d’environ 19,0 nm ± 0,1 nm. Cette valeur, plus importante que celles observées pour des monocristaux d’apatite au strontium ou au baryum dopés aux ions Nd3+ [44,45], peut être interprétée en termes de multitude de sites, due probablement à une plus grande modification de l’environnement local des ions Nd3+ dans la structure étudiée.
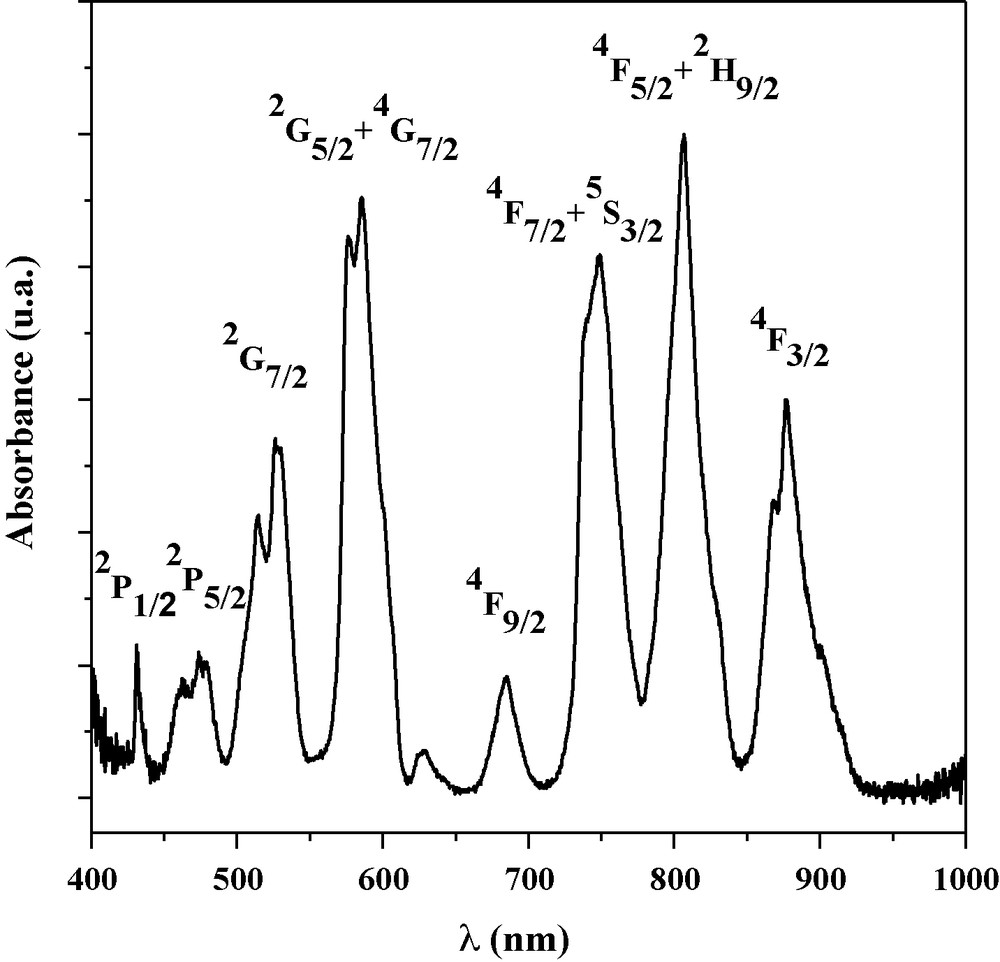
Spectre d’absorption optique de l’échantillon Sr9,8Nd0,2(PO4)5,8(SiO4)0,2F2.
Les spectres d’absorption des échantillons pour 0,2 < x ≤ 2,5, normalisés par rapport à l’intensité de la bande observée à environ 807 nm, sont montrés sur la Fig. 8. Certaines de leurs bandes présentent une structuration, probablement due à la multiplicité Stark [46]. Notons que la structuration de ces bandes montre un changement en fonction de la composition, correspondant à une augmentation (↑) ou diminution (↓) de l’absorbance relative à certaines lignes structurales (Fig. 8). Rappelons que les mesures d’absorption des ions activateurs sont sensibles aux changements structuraux de la matrice hôte. Ce changement serait dû, dans ce cas, à la multiplication des environnements locaux des ions Nd3+ au fur et à mesure que leur teneur augmente. Cela est en accord avec les résultats de diffusion Raman qui montrent qu’il y a multiplication des environnements structuraux des ions Nd3+ avec l’augmentation de leur quantité.

Spectres d’absorption optique des britholites de formule Sr10-xNdx(PO4)6-x(SiO4)xF2 avec 0,2 ≤ x ≤ 2,5.
3.5 Spectroscopie d’émission
Les mesures de luminescence ont été effectuées dans la gamme spectrale 1000–1400 nm. Sur la Fig. 9 est présenté le spectre de l’échantillon pour x = 0,02. Ce spectre comporte deux bandes d’émission centrées à 1054 et 1324 nm. Elles sont respectivement attribuées aux transitions 4F3/2 → 4I11/2 et 4F3/2 → 4I13/2. Parmi ces bandes, celle associée à la transition électronique 4F3/2→4I11/2 est la plus intense ; la valeur de sa largeur effective Δλeff est d’environ 19,5 ± 0,1 nm. En plus des raies caractéristiques de l’ion Nd3+, le spectre comporte une raie supplémentaire centrée autour de 1160 nm. Elle figure aussi sur le spectre de l’échantillon non substitué. (Fig. 9 en trait interrompu). Elle serait due à un centre luminescent présent en tant qu’impureté.

Spectre de luminescence des échantillons Sr10(PO4)6F2 et Sr9,98Nd0,02(PO4)5,98(SiO4)0,02F2 (T = 300 K ; λexc = 514,5 nm).
Sur la Fig. 10 sont montrés les spectres obtenus pour les échantillons pour x = 0,02, 0,20 et 2,00. Ils ont été normalisés par rapport à l’intensité de la bande observée à λp = 1054 nm dont l’élargissement croît avec l’augmentation de x. Cet élargissement est plus prononcé du coté des basses énergies (λ > λp). L’encart de la Fig. 10 présente la variation de sa largeur effective Δλeff en fonction de x. Comme le montre ce graphique, la largeur croît de 19,7 ± 0,1 nm à 34,0 ± 0,1 nm lorsque x varie de 0,02 à 2,00. Notons que l’élargissement s’accentue au-delà de x = 0,20.
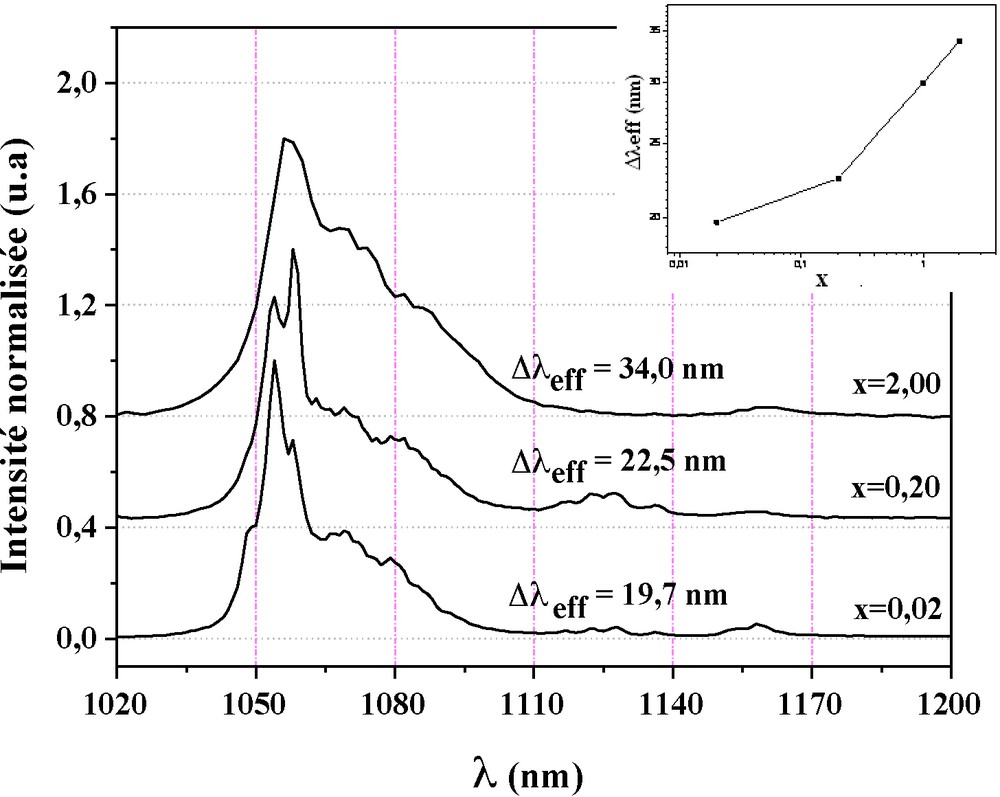
Spectres de luminescence des britholites de formule Sr10-xNdx(PO4)6-x(SiO4)xF2 avec x = 0,02, 0,20 et 2,00.
Du point de vue structural, l’élargissement des bandes d’émission peut être interprété en termes d’une nouvelle distribution des environnements locaux des ions Nd3+ correspondant à de nouveaux sites spectroscopiques dont le nombre se trouve supérieur à celui des sites cristallographiques théoriques. Pour certains composés, il peut atteindre 11 sites spectroscopiques inéquivalents [47].
4 Conclusion
L’objectif de ce travail a été la préparation par réaction à l’état solide de fluorbritholites strontiques contenant du néodyme de formule générale Sr10-xNdx(PO4)6-x(SiO4)xF2 avec 0 ≤ x ≤ 6. Dans le but d’étudier l’effet de l’incorporation de la paire d’ions Nd3+ et SiO44− sur la structure de l’apatite, les produits obtenus ont été caractérisés par différentes techniques.
L’étude par DRX a montré que l’obtention d’une phase apatitique pure n’est possible que pour x ≤ 2,5. Au-delà de cette valeur il y a, en plus, formation des phases secondaires Sr2SiO4 et Nd2SiO5. La variation des paramètres de maille en fonction de x a montré qu’il y a formation d’une solution solide discontinue de part et d’autre de x = 3. L’analyse par spectroscopies Raman, IR et absorption optique montre l’existence d’une multiplication des environnements locaux des ions Nd3+, qui crée dans la structure de l’apatite un certain désordre dont l’amplitude augmente avec x. Les mesures de luminescence ont montré que ces environnements correspondent à de nouveaux sites spectroscopiques.