

1 Introduction
The ability of tin (IV) to form compounds with coordination numbers greater than 4 through donor-acceptor interactions with Lewis bases has been largely explored over the last decades. The ensuing hypervalent derivatives have attracted considerable interest as models for the intermediate/transition-state structures of nucleophilic substitutions at tetracoordinate group 14 centers [1]. Most of these stannane adducts involve hard N- or O-based donors, and comparatively, little is known with soft phosphine donors. About ten P-adducts have been structurally authenticated with SnX4 and RSnX3 compounds (X = halogen) [2], but only two (adducts I and II) with the less Lewis acidic R3SnX moieties are currently known (Fig. 1) [3,4], High-field shifts of the 119Sn NMR resonance signals compared to the related tetracoordinated tin compounds and short PSn distances are diagnostic for the presence of P→Sn interactions that are favored in both compounds by rather flexible linkers, resulting in unstrained 5-membered ring structures.

Adducts I and II.
Our interest in the chemistry of ambiphilic compounds [5,6] has led us to synthesize main group compounds combining both donor and acceptor groups. As part of this effort, we observed that phosphine-boranes based on the ortho-phenylene backbone (type A, Fig. 2) sometimes display an intramolecular P−B interaction leading to the formation of a strained four membered-ring (Fig. 2) [7].
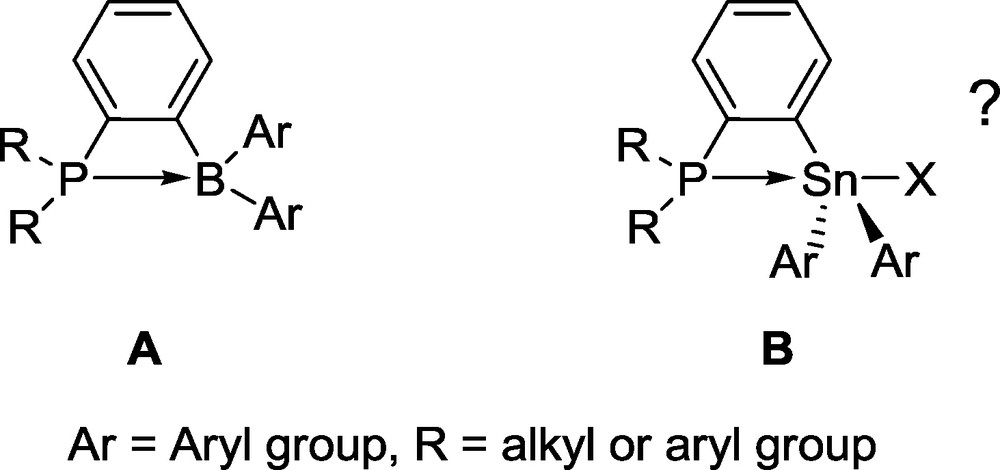
Type A phosphine-boranes.
In this article, we seek to determine if related donor-acceptor interactions could be observed in ortho-phenylene-based phosphine-stannane derivatives (type B, Fig. 2), despite the weaker Lewis acidity of the tin(IV) derivatives vs. boranes (Fig. 3).
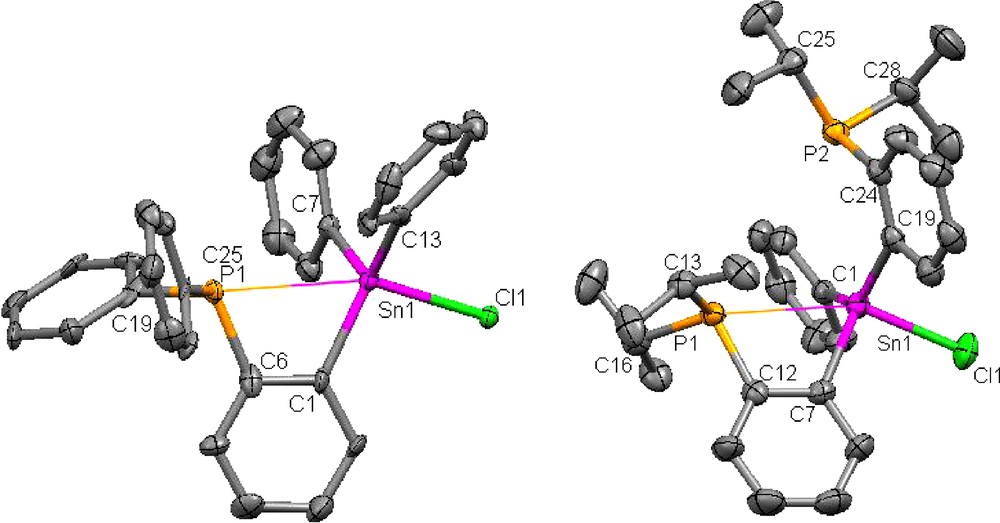
Crystal structure of 1 (left) and 2 (right) (H-atom omitted for clarity, ellipsoids drawn at 50% probability). Selected bond lengths(Å) and angles(°) for 1 are as follows: Sn(1)-C(13) 2.122(16), Sn(1)-C(1) 2.131(16), Sn(1)-C(7) 2.132(13), Sn(1)-Cl(1) 2.512(3), Sn(1)-P(1) 3.125(4), P(1)-C(19) 1.816(15), P(1)-C(6) 1.850(18), P(1)-C(25) 1.861(14), C(13)-Sn(1)-C(1) 118.8(6), C(13)-Sn(1)-C(7) 117.2(5), C(1)-Sn(1)-C(7) 114.4(5), C(13)-Sn(1)-Cl(1) 99.2(4), C(1)-Sn(1)-Cl(1) 101.1(4), C(7)-Sn(1)-Cl(1) 101.1(4), C(19)-P(1)-C(6) 104.8(7), C(19)-P(1)-C(25) 103.7(6), C(6)-P(1)-C(25) 105.3(7), C(1)-C(6)-P(1) 113.1(13), C(6)-C(1)-Sn(1) 117.9(12), P(1)-Sn(1)-Cl(1) 159.7(1). Selected bond lengths(Å) and angles(°) for 2 are as follows: C(1)-Sn(1) 2.132(4), C(7)-Sn(1) 2.155(4), C(19)-Sn(1) 2.153(4), Cl(1)-Sn(1) 2.4499(10), C(12)-P(1) 1.832(4), C(13)-P(1) 1.865(4), C(16)-P(1) 1.855(4), C(24)-P(2) 1.833(4), C(25)-P(2) 1.855(4), C(28)-P(2) 1.881(4), P(1)-Sn(1) 3.120(1), P(2)-Sn(1) 3.528(1), C(12)-C(7)-Sn(1) 117.1(3), C(7)-C(12)-P(1) 113.5(3), C(1)-Sn(1)-C(7) 114.98(13), C(1)-Sn(1)-C(19) 134.42(13), C(7)-Sn(1)-C(19) 106.91(13), C(1)-Sn(1)-Cl(1) 96.31(10), C(7)-Sn(1)-Cl(1) 96.08(10), C(19)-Sn(1)-Cl(1) 96.35(10), C(12)-P(1)-C(16) 103.80(17), C(12)-P(1)-C(13) 105.13(18), C(16)-P(1)-C(13) 104.16(17), P(1)-Sn(1)-Cl(1) 153.92(3).
2 Results and discussion
Reaction of ortho-(Ph2P)C6H4Li [6d,8] with Ph2SnCl2 in Et2O afforded ortho-(Ph2P)C6H4(SnPh2Cl) (1) which was isolated as a colorless solid in 29% yield. This compound has been characterized by multinuclear NMR spectroscopy and elemental analysis. The phosphorus center gives rise to a 31P NMR resonance at −1.0 ppm (JSn−P = 18.1 Hz) which can be compared to the value of −4 ppm observed for Ph3P. Interestingly, the 119Sn NMR resonance appears as a singlet at −101.7 ppm, a chemical shift which is distinctly upfield from that observed for Ph3SnCl (−45 ppm) [9]. This situation is reminiscent of that encountered in the known ortho-(i-Pr2P)C6H4)2SnPhCl (2) [10] which gives rise to a 119Sn NMR resonance at −126.4 ppm. In fact, the 119Sn NMR chemical shifts of 1 and 2 approach those reported for hypervalent tin species adducts such as [2-(Me2NCH2)C6H4]SnPh2Cl (-176.9 ppm) [11] or ClPh3Sn-pyridine (−219 ppm) [12]. These spectroscopic observations can be reconciled by invoking the presence of an intramolecular P→Sn interaction in 1 and 2. Because the rigidity of the ortho-phenylene backbone as well as the diverging orientation of the phosphine and stannane groups in 1 and 2 are not necessarily consistent with the formation of such P→Sn dative interactions, we resorted to X-ray crystallography.
Examination of the structure of 1 indicates that the P(1) and Sn(1) atoms are separated by 3.125(4) Å. This separation, which is well within the sum of the Van der Waals radii of the two elements (4.2 Å) [13], is commensurate with the presence of a dative interaction. In line with this argument, we note that the C(6)-C(1)-Sn(1) (117.9(1)°) and C(1)-C(6)-P(1) (113.1(1)°) angles are both smaller than 120°. The P(1)-Sn(1)-Cl(1) angle (159.7(1)°) and the sum of the Ceq-Sn(1)-Ceq angles of 350.3° suggest that the coordination geometry at tin is best described as trigonal bipyramidal. Altogether, the structure of 1 is reminiscent of that of compound I which possesses an intramolecular P→Sn dative interaction of 3.079(2) Å. The structure of 2 has also been determined. Inspection of the structure indicates that the P(1)-Sn(1) distance (3.120(1) Å) is very similar to that observed in 1. The second phosphorus atom remains far away from tin (P(2)-Sn(1) = 3.528(1) Å). This dichotomy can be assigned to the presence of a P(1)-Sn(1) dative interaction which closely resembles that observed in 1. This resemblance can be further ascertained by the similarity of the C(12)-C(7)-Sn(1) (117.1(3)°) and C(7)-C(12)-P(1) (113.5(3)°) and P(1)-Sn(1)-Cl(1) angle (153.92(3)°). The sum of the Ceq-Sn(1)-Ceq angles of 356.31° is also close to that observed in 1.
In order to better understand the nature of the P-Sn interaction present in these compounds, their gas phase structures have been optimized using Density Functional Theory (DFT) methods (functional: BP86 [14]; mixed basis set: P, Sn, Cl: Stuttgart RLC ECP [15]; F: 6–31 g(d’); C, H: 6–31 g(d)). The optimized geometries, which are close to those experimentally determined (Table 1), were subjected to natural bond orbital (NBO) analyses which identified a lp(P)→p(Sn) donor-acceptor interaction (Fig. 4). Moreover, a deletion calculation carried out by zeroing the Kohn-Sham matrix elements corresponding to the lp(P)→p(Sn) interaction in 1 and 2 shows that this interaction contributes to the stability of the molecule by Edel(P→Sn) = 18.9 kcal/mol in the case of 1 and Edel(P→Sn) = 24.4 kcal/mol in the case of 2 (Fig. 4). In turn, the tin center of these derivatives experiences significant electron donation from the phosphorus. The difference in Edel(P→Sn) between 1 and 2 is indicative of the increased donicity of the di(iso-propyl)phosphine moiety of 2. This view is confirmed by a computational study of the model compound ortho-(i-Pr2P)C6H4(SnPh2Cl) (3), the di(iso-propyl)phosphine analog of 1, which affords Edel (3) = 22.8 kcal. The donor-acceptor interaction between the phosphorus and tin atoms of these compounds was further ascertained by application of the Atom-In-Molecule (AIM) method [16]. This method, which allows for straightforward topological analysis of the electron density of a given compound, confirmed the presence of a bond path connecting the tin and phosphorus atoms of 1, 2 and 3. The values of the density at the P-Sn bond critical point of 1, 2 and 3 are respectively equal to 0.017, 0.019 and 0.018 e/bohr3. Although these values are very close to one another, the higher densities observed in the case of 2 and 3 can also be correlated to the increased donicity of the di(iso-propyl)phosphine moiety.
Experimental and computed bond lengths (Å) and angles (°) for 1–3.
| 1 X-Ray | 1 Density Functional Theory | 2 X-Ray | 2 Density Functional Theory | 3 Density Functional Theory | |
| P1-Sn | 3.125(4) | 3.294 | 3.1206(9) | 3.235 | 3.231 |
| P2-Sn | – | – | 3.528(1) | 3.601 | – |
| Sn-Cl | 2.512(3) | 2.505 | 2.450(1) | 2.536 | 2.511 |
| Sn-C-C | 117.9(1) | 118.6 | 117.1(3) | 117.7 | 117.4 |
| P-C-C | 113.1(1) | 115.9 | 113.3(3) | 114.7 | 115.1 |
| P-Sn-Cl | 159.7(1) | 155.2 | 153.9(1) | 153.3 | 156.3 |
| ∑ Ceq-Sn-Ceq | 350.32 | 349.8 | 356.31 | 356.4 | 350.6 |
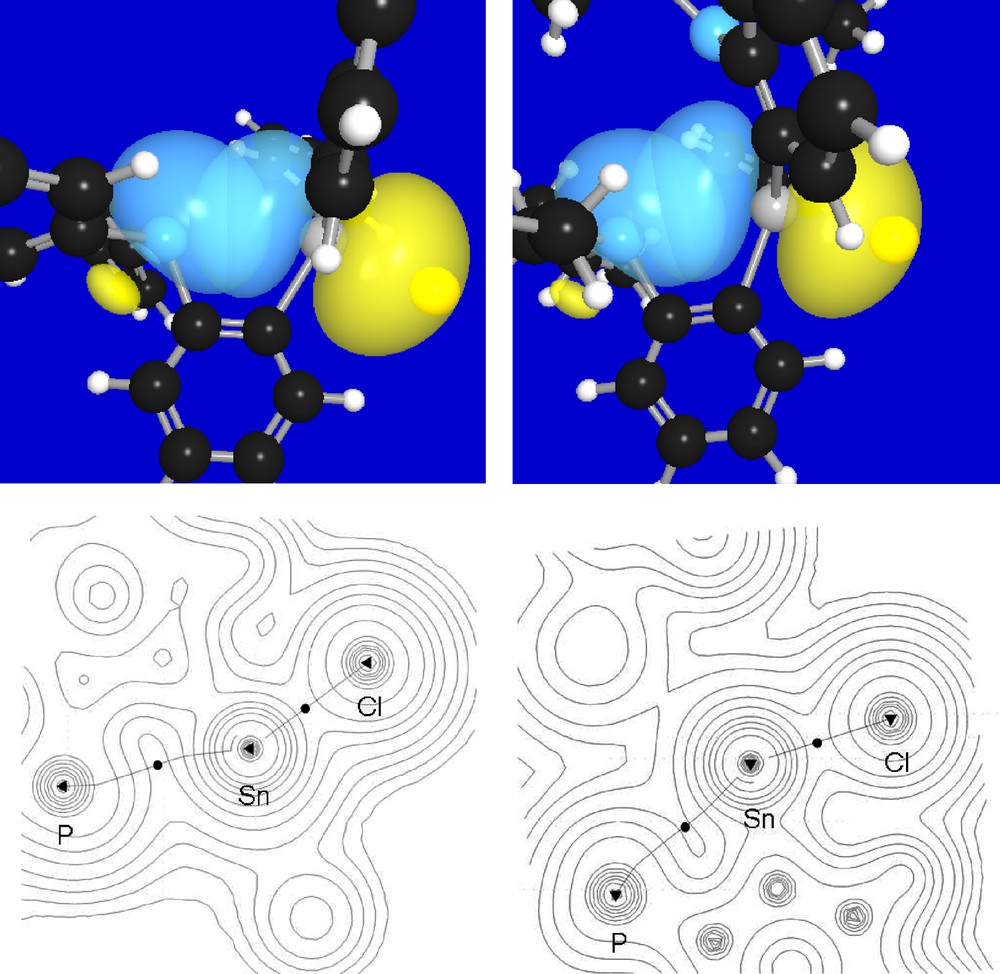
Top: NBO contour plots showing the lp(P)→p(Sn) donor-acceptor interactions involved in 1 (left) and 2 (right). Bottom: Atom-In-Molecule electron density maps for 1 and 2 in the P-Sn-Cl plane showing the relevant bond paths and bond critical points.
3 Conclusion
The results reported here evidence that, despite the rigidity of the ortho-phenylene backbone, the phosphine and stannane groups of 1 and 2 are engaged in an intramolecular P→Sn dative interaction.
4 Experimental section
4.1 General considerations
Ph2SnCl2 and n-BuLi were purchased from Aldrich. ortho-(Ph2P)C6H4Li 1 [6d,8] and 2 [10] was prepared according to the reported procedures. Solvents were dried by passing through an alumina column (CH2Cl2) or refluxing under N2 over Na/K (Et2O and n-hexane). NMR spectra were recorded on Varian Unity Inova 400 FT NMR (399.59 MHz for 1H, 161.75 MHz for 31P, 149.00 MHz for 119Sn) spectrometers at ambient temperature. Chemical shifts are given in ppm, and are referenced to residual 1H and 13C solvent signals and external H3PO4 (31P) and SnMe4 (119Sn).
4.2 Synthesis of compound 1
To a 15 mL Et2O solution of 2-bromophenyl-diphenylphosphine (1250 mg, 3.66 mmol) was added a n-hexane solution of n-BuLi (2.2 M, 1.83 mL, 4.03 mmol) at 0 °C. After stirring at room temperature for 20 min, this mixture was transferred into a 5 mL Et2O solution of Ph2SnCl2 (1.39 g, 4.03 mmol) at −78 °C. The reaction was allowed to warm up slowly (in 1 h) and stir for overnight. Next day, the solvent was removed under reduced pressure. The product was extracted with CH2Cl2 (3 × 10 mL), and the solution was filtrated through celite. After evaporating CH2Cl2, the resulting white solid was washed with hexane (3 × 10 mL) to afford 1 as crude product (0.60 g, 28.7% yield). Further recrystallization in CH2Cl2 at −30 °C afforded colorless crystals of 1 suitable for crystallographic measurements. 1H NMR (CDCl3) δ = 7.05 (t, 4H, 3JH−H = 8.5 Hz), 7.18–7.36 (m, 15H), 7.74 (d, 4H, 3JH−H = 7.8 Hz), 8.24 (d, 1H, 3JH−H = 7.6 Hz). 31P{1H} NMR (CDCl3) δ = −1.0 (3JSn−P = 18.1 Hz). 119Sn{1H} NMR (CDCl3) δ = −101.7 (bs).
4.3 X-ray measurements
The crystallographic measurements were performed at 110(2) K for 1 and 133(2) K for 2 using a Bruker APEX-II CCD area detector diffractometer (Mo-Kα radiation). A specimen of suitable size and quality was selected and mounted onto a nylon loop for 1 and glass fiber for 2. Semi-empirical absorption corrections were employed [17]. The structures were solved by direct methods (SHELXS-97 [18] or SHELXTL [19]) and refined using the least-squares method on F2 [20].
Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC–788735 (1) and 788736 (2). These data can be obtained free of charge via www.ccdc.cam.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223_336_033; or deposit@ccdc.cam.ac.uk).
4.4 Computational details
DFT calculations were performed using Gaussian program [21]. Structural optimizations of 1–3 were carried out using BP86 functional. C, H were treated with 6-31G(d) basis set; F were treated with 6-31G(d’) basis set; P, Cl, Sn were treated with Stuttgart RLC ECP basis set [15]. NBO calculations were carried out using Gaussian program with the same functional and basis set. The topology of the electron density of 1–3 was obtained by performing a single point calculation on the optimized structure using different basis set (6-31G(d) for C, H; 6-31G(d’) for F; 6-31G+(d) for P, Cl; cc-PVDZ-pp for Sn). AIM analysis was carried out in AIM2000 program [22].
Acknowledgments
This work was supported by NSF (CHE-0646916), the Welch Foundation (A-1423), the CNRS, the Université de Toulouse and the ANR (ANR-06-BLAN-0034). FG thanks the Laboratory for Molecular Simulation at Texas A&M University for providing computing resources, and DB thanks the COST action CM0802 PhoSciNet for supporting this work.