

1 Introduction
Supported palladium catalysts display excellent activity towards hydrogenation of aromatics compounds and have also been reported in the literature as the catalyst of choice in benzene hydrogenation [1–5]. Many parameters determine its catalytic activity in hydrogenation processes. The catalytic activity strongly depends on the metal loading, nature of metal precursors, preparation method, metal particle size and reduction temperature [3,6–11]. The method of preparation plays a crucial role in the catalytic behavior of these systems. Conventional preparation consists in the calcinations in air of supported precursors then activation in hydrogen of the metal oxides obtained. A study of support effects in nitrobenzene hydrogenation showed that Pd/Mg–Al catalysts were more active than Pd/MgO and Pd/Al2O3 catalysts due to a better metal dispersion [10]. The activity decreased with the metal loading. In same reaction, PdCl2 as a metal precursor was found more active than Pd(NO3)2 or Pd(OOCCH3)2 [6]. Support effects in liquid phase hydrogenation of benzene were also evidenced. Pd/CNT catalysts exhibited higher activity than Pd supported on zeolite and Pd supported on activated carbon [9]. Variation in metal loading in the range 2–12% led to minimum activity for 8%. The effect of the reduction temperature on the specific rate in benzene hydrogenation was studied for Pd/SiO2 catalysts [8]. The results obtained show that the specific activity decreased with increasing reduction temperature.
More recently, new methods were explored in order to improve metal dispersion surface properties, notably the chemical reduction methods. The Pd/SiO2 catalysts obtained from the reduction of an organometallic precursor with dihydrogen showed higher activity in benzene hydrogenation [1]. Several authors prepared palladium nanoparticles by reduction of the palladium salts by a reducing agent like hydrazine, alcohols, KBH4, polyols, microwave irradiation and γ-radiolysis [12–16]. The Pd/SiO2 catalysts were prepared by ethanol as reducing agent and PdCl2 as precursor exhibited a good activity in benzene hydrogenation [3,4]. The oxidation after reduction by ethanol improves the catalytic performances of these catalysts. Pd/Nb2O5 catalysts with a low dispersion have been prepared by the reduction of palladium precursors by ethylene-glycol or hydrazine [17]. They show a good reactivity and selectivity in hexa-1,5-diene hydrogenation. Pd/Al2O3 catalysts were prepared by radiolysis method in aqueous medium [16]. They exhibit higher dispersion and activity in hydrodechlorination (HDC) of chlorobenzene, as compared to the catalysts prepared by the conventional calcination/hydrogen reduction method. Pd/Nb2O2 and Pd/Al2O3 catalysts prepared by microwave irradiation show low dispersion, compared to the conventional catalysts [15,18]. However, these catalysts exhibited higher activity and stability during the HDC reactions than that the conventional catalysts. The Pd-B/SiO2 catalysts were prepared by reduction of PdCl2 as precursor by KBH4 as reducing agent [14]. The results obtained show that these catalysts are more active in hydrogenation of nitrobenzene than the conventional catalysts.
In previous work, we prepared supported nickel catalysts by the hydrazine method using nickel acetate as the metal precursor. Metal particles of about 3 nm in size or less could be obtained as well as various morphologies (spherical, whiskers-like or chestnut bur-like), depending on the conditions of preparation (temperature, Ni2+/hydrazine ratio, reactants concentration) [19], nature of the support [20,21] or presence of a second metal [22,23]. The difference in the particle morphology was ascribed to differences in the number of nuclei formed or/and the growth rate during the reduction by the hydrazine. The activity and stability of the nickel phase obtained in the gas phase hydrogenation of benzene depended on the nanoparticles size and shape. It was found that the activity of the hydrazine catalysts could be up to three times higher than that of conventional catalysts.
In this work, we present results obtained with palladium based catalysts prepared by the hydrazine method using the PdCl2 and silica as a precursor and a support, respectively. The metal loading was in the range 1–10% (wt.%). The structure and morphology of these Pd catalysts have been studied by using X-Ray Diffraction (XRD), Transmission Electronic Microscopy (TEM), Electron Diffraction (ED) and Energy Dispersive X-Ray (EDX) analysis. The catalysts were also tested in the gas phase hydrogenation of benzene.
2 Experimental
2.1 Catalyst preparation
Doubly distilled water was used as solvent. Palladium chloride (Fluka), aqueous hydrazine 24–26% (Fluka) and the silica support (Sigma) of 200 m2 g−1 and grains of 0.014 μm were used as received.
The silica support (5.0 g) was impregnated with 80 mL of aqueous solution of the precursor salt. The palladium chloride concentration in the solution was calculated to obtain the nominal composition of 1.0–10.0%Pd w/w. The suspension was stirred for 36 h at room temperature. The solvent was then evaporated at 80 °C under vacuum and the obtained solid dried for 16 h at 110 °C.
The preparation of the supported palladium particles hydrazine catalysts was performed under argon atmosphere in a three-necked reaction flask of 250 mL plunged in a water bath. The supported palladium chloride precursor was introduced in the reaction flask filled with 100 mL of bidistilled water. The suspension obtained was stirred for 20 min at room temperature then 1–2 mL of hydrazine solution (in excess) was added. The suspension changed to dark brown color. The dark brown suspension was maintained for 20 min at room temperature. The pH was 8–10 and remained almost constant during the reduction process. The suspension was filtered, washed several times with ethanol. The filtrate was dried under vacuum. The resulting solid was stored under argon. The samples were denoted as PdSiO2X where X is the metal content.
The degree of reduction of Pd+2 ions has been estimated by measurement of the amount of nitrogen evolved during the reaction of reduction, assuming the following chemical equation:
| Pd+2 + 1/2N2H4 + 2OH− = Pd0 + 1/2N2↑ + 2H2O |
A classical supported catalyst, denoted as PdSiO21(C), was prepared by calcinating the supported palladium chloride (1% Pd) in air at 300 °C for 2 h.
The prepared catalysts are listed in Table 1.
H2-chemisorption and TPD experiments for the Pd/SiO2 catalysts.
| Catalyst | Pd loading (wt%) | H2 adsorbed (μmol gPd−1) | Metal areaa (m2 gPd−1) | H2 desorbed (μmol gPd−1) | Pd particle size (nm) | |
| XRD | H2 chemisorption | |||||
| PdSiO21 | 1.0 | 614.2 | 55.6 | 5916.0 | 6.8 | 8.9 |
| PdSiO23 | 3.1 | 454.7 | 41.2 | 3981.3 | 11.0 | 12.1 |
| PdSiO25 | 5.0 | 224.3 | 20.3 | 1981.5 | 22.6 | 24.5 |
| PdSiO210 | 10.0 | 184.7 | 16.7 | 1694.7 | 28.4 | 29.8 |
| PdSiO2 (C) | 1.0 | 2810.0 | 254.5 | 3557.2 | – | 1.9 |
a Calculated from H2 chemisorption.
2.2 Catalyst characterization
The palladium composition of the catalysts was determined on a Varian AA1275 atomic absorption spectrophotometer. The XRD patterns I(θ) were recorded at a classical θ/2θ diffractometer using Cu Kα radiation.
TEM images were obtained on a Philips CM20 STEM microscope equipped with Energy Dispersive X-Ray (EDX). Samples were dispersed in ethanol by sonication and dropped on a copper grid coated with a carbon film.
Chemisorption experiments for both hydrazine and calcined catalysts were carried out with a sample of 100 mg on a pulse chromatographic microreactor equipped with the catharometric detector of a microchromatograph (AT M200, Hewlett Packard) fitted with molecular sieve columns and MTI software. The sample was pretreated with pure H2 with a flow rate of 50 mL min−1 for 2 h at 300 °C, using a heating rate of 10 °C min−1. It was then purged under argon atmosphere for 2 h with a flow rate of 100 mL min−1 and cooled to 70 °C. After that, gaseous hydrogen (100 ppm in argon) was injected in the reactor every 2 min until saturation of the sample at 70 °C in order to avoid β-hydride formation [24]. The hydrogen saturated sample was purged at the same temperature under argon for 1 h, a time after which no hydrogen (< 1 ppm) was detected in the exit gas. It was subsequently submitted to a TPD under same gas (50 mL min−1) with a heating rate of 7.5 °C min−1 up to 800 °C.
H2 chemisorption was applied to measure the accessible Pd surface using Pds/Ha = 1 for adsorption stoichiometry [25]. The metal surface area (S, m2 gPd−1) was calculated from H2 uptake using Eq. (1):
| (1) |
| (2) |
2.3 Catalyst testings
The gas-phase hydrogenation reaction was carried out in a quartz fixed-bed reactor after activation under hydrogen as described above for chemisorption studies. The catalytic tests were conducted with a sample of 100 mg and a feed gas of 1% C6H6/H2 and total rate flow of 50 mL min−1. The reactant and product analysis were conducted on line using a Hewlett-Packard 5730A FID gas chromatograph equipped with TCEP (2 m, 1/8 in) and Sterling (3 m, 1/8 in) columns.
The utilized gases were purchased from Air Liquide. Oxygen traces were eliminated from argon (99.995%) and hydrogen (99.995%) by using a manganese oxytrap (Engelhardt) whereas helium (99.999%) was used as received.
3 Results and discussion
3.1 XRD and TEM studies
The XRD patterns of the Pd/SiO2 catalysts are shown in Fig. 1. All samples exhibited a broad peak around 2θ = 21.3° which is due to the amorphous silica. The fresh or H2/300 °C treated hydrazine catalysts showed a large main band at 2θ = 40.1°, characteristic of metallic palladium phase of fcc structure. The peaks at 40.1°, 46.6° and 68.1° can be assigned to (111), (200) and (220) crystalline plane diffraction, respectively. The particle size of hydrazine catalysts, as estimated using the Debye-Sherrer equation, increased from 6.8 to 28.4 nm with increasing palladium composition (Table 1). Moreover, XRD spectra of the catalysts before and after the treatment under hydrogen at 300 °C/2 h show significant change in the crystallite.

XRD patterns of the palladium catalysts: (a) PdSiO21 (C); (b) PdSiO23; (c) PdSiO25.
Variations in the metal particle size may be ascribed to metal-support interaction. Indeed, for high palladium content, part of the precursor does not directly interact or interacts less directly with the support and will probably migrate more easily, accounting for bigger average particle size during the reduction process. In contrast, for lower palladium content, the metal particles were rather stabilized by the support and less prone to coalescence. Particle size increase may also be due to redissolution/precipitation of the metal precursor during the hydrazine reduction process. In such a case, the reduction partly occurs in the solution or in the vicinity of the support.
Classical PdSiO21(C) exhibited a flat XRD spectrum as a result of the existence of very small particles, probably subnanoparticles (Fig. 1). Metal dispersion is most probably determined during the calcination step during which small PdOx particles were stabilized on the support. The subsequent reduction by gaseous hydrogen at 300 °C did not change their morphology. This is in contrast with the larger particles (6.8 nm) observed for the hydrazine catalyst PdSiO21. In this case, the metal salt precursor, not in strong interaction with the support, allowed coalescence of the Pd° particle formed during the reduction process. Pd/SiO2 catalysts prepared by ethanol as reducing agent exhibited an enhanced activity when the metal nanoparticles obtained are subsequently calcined in air then activated in hydrogen atmosphere [3,4]. This was attributed to the morphological changes on Pd particles during the calcinations step.
TEM micrographs, EDX spectrum and ED pattern of Pd/SiO21 sample were performed. Small Pd particles are well evidenced (Fig. 2a). The size distribution is between 3–9 nm, and most of them are located at around 6 nm. These results are in good accordance with particle size values estimated by XRD (Table 1). The ED pattern (Fig. 2b) confirmed the fcc structure of the palladium particles. The EDX analysis (Fig. 2c) indicated the presence of metallic palladium on the support with content which well agrees with that of the starting material. In contrast, no residual chlorine was detected on the catalyst surface.
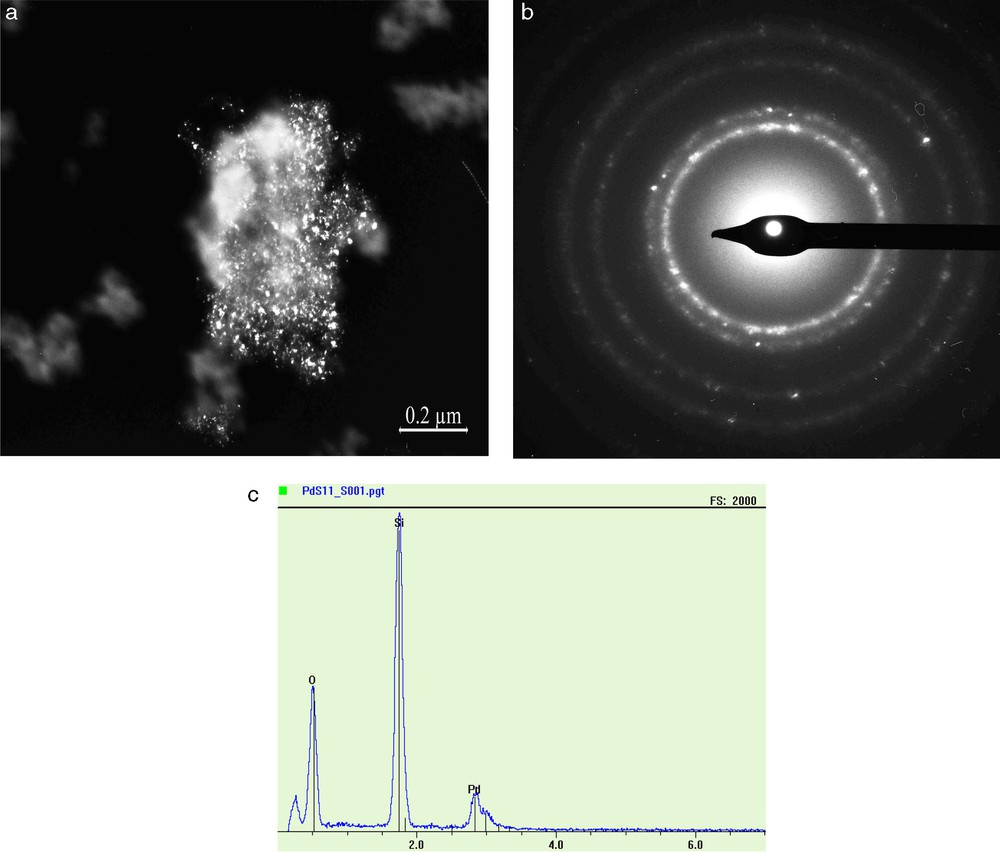
(a) TEM images of the PdSiO21 catalyst; (b) ED pattern; (c) EDX analysis for TEM image in (a).
3.2 H2-adsorption studies
Several studies on supported spalladium catalysts howed that the metal dispersion depended on many factors like metal loading, nature of precursor, treatment temperature and nature and surface area of support [25,26]. Metal dispersion in conventional palladium catalysts is generally determined by CO chemisorption [3,11,16,27]. Hydrogen chemisorption has also been used taking into account possible formation of palladium hydride [28–31]. Indeed, β-PdH species are formed during H2 chemisorption at room temperature of Pd supported catalysts prepared by calcination and hydrogen activation of a metal precursor [28–32]. It was shown that this hydride is unstable and its decomposition appears between 27–300 °C [6,10,31–35]. A number of publications have reported that formation of Pd hydride depends on the particle size of palladium; larger Pd particles more easily form Pd hydride whereas smaller Pd particles may be unable to form the Pd β-hydride phase [28,33]. It is also admitted that chemisorption carried out at 70 °C avoid β-PdH formation [24,36,37]. In contrast, the Pd hydride was not evidenced in case of non-conventional catalysts. This was claimed for Pd catalysts prepared by microwave heating [15] and radiolysis method [16] were observed.
Taking into account these considerations, we determined the metal surface area by the H2-adorption method at 70 °C for both hydrazine and conventional catalysts. Calculation were performed using Pds/Ha = 1 for adsorption stoichiometry [25]. Discussion on the nature of the hydrogen species adsorbed is reported in the next section.
The amount of hydrogen adsorbed depends on the palladium loading for hydrazine catalysts: it sharply decreases when the palladium composition increases from 1% to 10%Pd, passing from 614.2 to 184.7 μmol gPd−1. This indicates an increase of the particle size with the metal loading. This is in fair accordance with the particle size determined by XRD. TEM (Fig. 2a) also confirmed the particle size for PdSiO21.
The conventional catalyst PdSiO21(C) adsorbed 4.5 times more hydrogen than the PdSiO21 hydrazine catalyst did: 2810.0 μmol gPd−1 versus 614.2 μmol gPd−1. This is ascribed to the higher dispersion of the palladium phase on silica; therefore the method of preparation has a significant effect on the metallic surface area. As showed the XRD study, calcination of the catalyst precursor favored a better dispersion of palladium than the direct reduction in the hydrazine media.
3.3 H2-TPD study
The H2-TPD can shed some light on differences in metal-support interaction and electronic properties of supported metal particles [38,39].
Inspection of H2-TPD for classical palladium based catalysts shows various profiles. Recently, the H2-TPD profile for Pd/SiO2 catalyst prepared by impregnation of Pd(NO3)2 and calcination at 500 °C shows a wide desorption peak at temperatures ranging from 25–350 °C and a peak of maximum rate of desorption at 480 °C [31]. These peaks indicate that several adsorption states exist. Low and high temperature peaks were attributed to weakly and strongly bound hydrogen on the catalyst surface. Peak at 200 °C is believed to be due to the decomposition of β-PdH species. A broad peak at 85 °C due to the weak adsorption of hydrogen was also evidenced in the H2-TPD profiles for Pd/SiO2 catalysts [35]. Similar trends are reported for palladium catalysts deposited on other supports. The H2-TPD profile for Pd/Al2O3 catalysts exhibits a wide desorption peak at the temperature ranging from 50 to 450 °C [40]. This peak is believed to represent hydrogen desorption as a continuum from the metal, metal-support interface and support. A main peak at 677 °C was observed during H2-TPD experiments on Pd/MgO catalysts [41]. This peak was attributed to the partial desorption of hydrogen that is chemisorbed on both palladium and support.
In the present work, the H2-TPD study was carried out after a hydrogen treatment at 300 °C for 2 h and hydrogen adsorption at 70 °C. The results of hydrogen desorption of hydrazine catalysts are illustrated in Fig. 3.

H2-TPD profiles of the supported palladium catalysts: (a) PdSiO210; (b) PdSiO25; (c) PdSiO23; (d) PdSiO21.
The hydrazine catalysts exhibited two peaks beginning from 350 °C at 450 and 540 °C respectively. They are attributed to hydrogen species strongly bonded to palladium or as spilt-over species [42–44]. Remarkably no peak appeared below 350 °C. This suggests that no β-PdH species were formed although the larger sized particles were obtained. This is in fair accordance with H2-TPR experiments (not reported). No negative peak corresponding to β-PdH decomposition was observed in the temperature range 25–750 °C. This is in good accordance with the absence of negative peak in the TPR patterns for non conventional catalysts prepared by microwave heating [15] or radiolysis method [16]. The authors concluded that no β-PdH species were formed due to the small size of the particles obtained [10,28,33]. Since the latter catalysts contains small sized Pd nanoparticles, one is allowed to think that the absence of β-PdH species palladium metal hydride rather than the presence of small metal particle size would be characteristic of palladium catalysts reduced at low temperatures without the help of gaseous hydrogen rather.
A quite different pattern is shown by the conventional catalyst PdSiO21(C) as reported in Fig. 4. The hydrogen desorption comprised two domains of temperature denoted type I and type II below and above 350 °C, respectively. In the first domain, two overlapping peaks were observed at 110 and 178 °C, respectively. In the second domain, a main peak with a shoulder appeared at 510 and 410 °C, respectively. Peaks of type I are attributed to hydrogen species weakly attached to the palladium phase. Peaks of type II are similar to that of hydrazine catalyst PdSiO21 with a shift to lower temperatures. They are also attributed to hydrogen species strongly bonded to palladium or spilt-over species [42–44], although to a lesser extent than for the hydrazine catalysts. The classical catalyst, in contrast to the hydrazine reduced catalysts, contained metal sites of weakly bonded H atoms which suggest a better activity in hydrogenation reactions.

H2-TPD profile of the conventional palladium catalyst PdSiO21 (C).
All catalysts desorbed more hydrogen than they adsorbed (Table 1). The catalysts behave as hydrogen reservoirs. The intensity of peaks increased with the decrease of the Pd loading. The amount of hydrogen desorbed increased from 1694.6 to 5915.3 μmol gPd−1 when the palladium content diminished from 10 to 1% Pd. There is, however, a small change in the amount of hydrogen desorbed between 5 and 10% Pd. In comparison, the conventional catalyst desorbed less (3557 μmol gPd−1) than the hydrazine homolog. For this catalyst, hydrogen desorption mainly comes from the high temperature domain.
The excess hydrogen shows that a part of the reactant molecules is strongly retained on the catalyst through the so-called spillover effect. The high temperature desorption would be due to the so-called spillover effect. This process occurred during the hydrogen activation for hydrazine catalysts and conventional catalyst (see Experimental).
Interestingly, the hydrogen reservoir increases with decreasing particle size for the hydrazine catalysts. It is well established that the intensity of the spillover effect (volume or the hydrogen reservoir) increases with the metal dispersion: the smaller the metal particles, the greater the number of active sites from which the H-adspecies diffuse to the support [43,44]. The small change in the amount of hydrogen desorbed between 5 and 10% Pd can be attributed to small change in the particle size.
3.4 Catalytic activity
The hydrogenation of aromatics is of major importance in the chemical industry because of the stringent environmental regulations governing their concentration in diesel fuels [45–46]. Benzene hydrogenation has been chosen as the model aromatic feedstock [47–48]. This reaction has also been used as model reaction in heterogeneous catalysis by metals where metal-support interactions are involved [47–50].
The test-reaction was carried out after a hydrogen treatment at 300 °C for 2 h for all catalysts. Activated conventional or non-conventional catalysts became active and selective in the gas phase hydrogenation of benzene. At all reaction temperatures only cyclohexane is found in the exit gas. Steady-state and stable activity was rapidly reached. Conversions were in the range 4–100%. The bare silica support, previously treated in aqueous hydrazine, was found inactive.
The catalysts exhibited a maximum of activity as a function of the reaction temperature (Fig. 5). This maximum was reached around 200–225 °C. It is attributed to a decrease of the surface coverage by benzene molecules at the higher temperatures.

Effect of palladium loading on the specific rate for hydrazine catalysts.
For hydrazine catalysts, the activity increases with decreasing Pd content, that is small particles are the most active. Table 2 reports detailed values of catalytic activity for the reaction temperature of 100 °C. It can be seen that he specific rate increases from 0.16 μmol min−1 gPd−1 for 10%Pd to 1.03 μmol min−1 gPd−1 for 1%Pd. The difference in activity is around 85%.
Catalytic activity and TOF for the Pd/SiO2 catalysts.
| Catalyst | Specific rate (× 103) (mol min−1 gPd−1) | TOF (× 102) (s−1) |
| 100 °C | 100 °C | |
| PdSiO21 | 1.03 | 1.40 |
| PdSiO23 | 0.54 | 1.00 |
| PdSiO25 | 0.27 | 1.01 |
| PdSiO210 | 0.16 | 0.74 |
| PdSiO2 (C) | 1.68 | 0.49 |
In comparison to hydrazine catalysts, the classical catalyst was much more active with a maximum conversion at a lower temperature (175 °C) and a specific rate 1.6–3.7 times higher below maximum conversion temperature (Fig. 6). High specific rate is attributed to higher dispersion of the Pd particles on the support (Table 1).
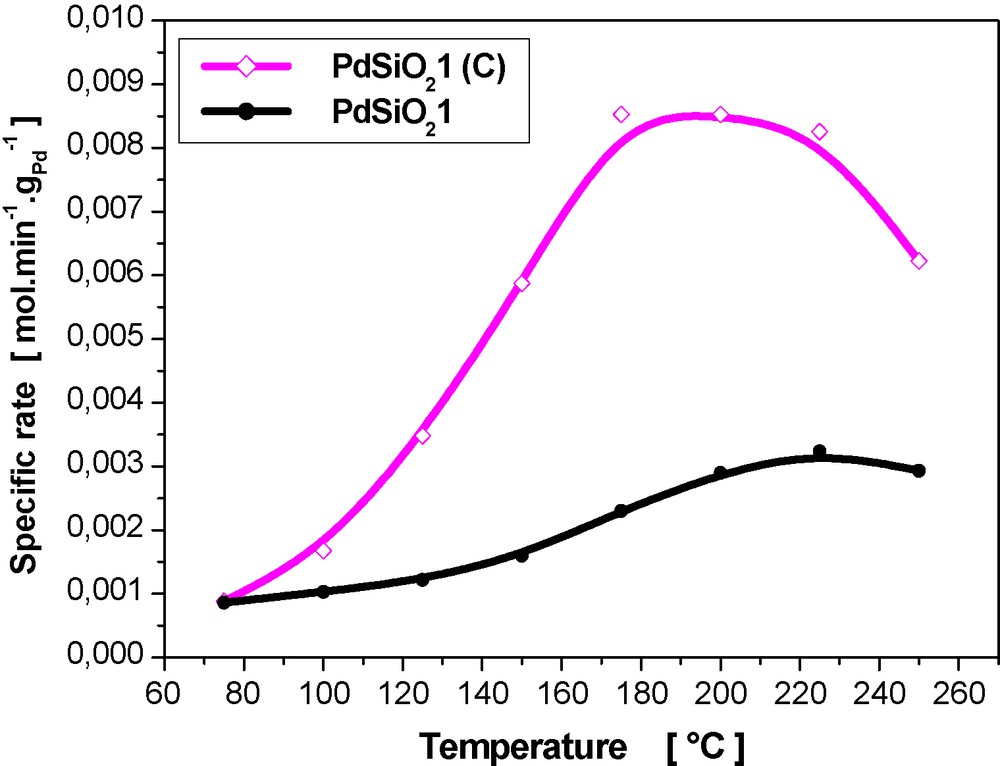
Specific rates in benzene hydrogenation over conventional PdSiO21 (C) and hydrazine PdSiO21 catalysts.
The use of turnover frequency (TOF) allows one to make a valuable comparison of the intrinsic activity of the palladium surface atoms. The results obtained are in the range of 0.0140–0.0074 s−1. This is in good accordance with TOF's published for benzene hydrogenation in gas phase [5,27].
Strikingly, lower TOF's are given by the classical catalyst PdSiO21(C). The intrinsic activity is lower in the whole range reaction temperature (Fig. 7), notably at lower temperatures. For example, the hydrazine catalysts give a value of 0.0049 s−1, three times higher than that of the conventional catalyst, for the reaction temperature of 100 °C (Table 2). These results are quite unexpected since the smallest Pd particles are less reactive than the largest. This reverse size effect has not been reported. Further work is needed to understand the contrasting results obtained. Note that, generally speaking, the benzene hydrogenation is considered as a structure insensitive reaction [8], although there are several contradictions in the literature regarding this problem [51–53].
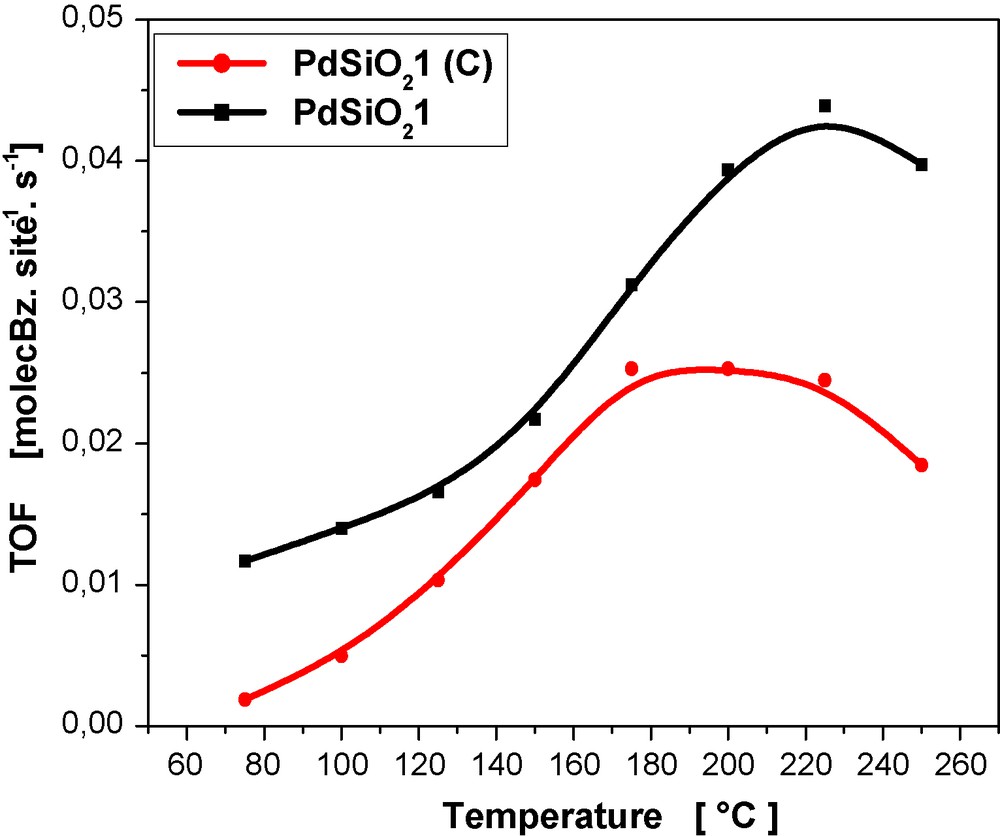
Intrinsic activity (TOF) in benzene hydrogenation over conventional PdSiO21 (C) and hydrazine PdSiO21 catalysts.
4 Conclusions
Metal particles with a size range 6.8–28.4 nm were obtained by reduction of SiO2-supported palladium chloride aqueous hydrazine. Dispersion, hydrogen storage and activity in benzene hydrogenation increased with decreasing particle size. In comparison, a 1%Pd classical catalyst, prepared by calcination then hydrogen reduction of same precursor, was found much more dispersed (mean particle size of 1.6 nm) and more active (specific rate 1.6–3.7 times higher) than the 1%Pd hydrazine catalyst. However, TOF's calculations indicated a greater reactivity of the metal surface atoms for the hydrazine catalyst. It also stored greater amounts of hydrogen according to the TPD experiments. Further studies are necessary to a better understanding of the results obtained.