

1 Introduction
The activation and functionalization of small, readily available, molecules represent important processes for the advancement of knowledge on chemical reactivity, which, in addition, may open the way to many industrial applications. This is particularly important at the present time, when more and more stringent legislation requires that industries devote increasing attention to the environmental impact of any newly implemented processes.
A huge number of studies have focused on the activation of simple molecules, either largely available in nature, such as CH4 [1,2], CO2 [3,4] and N2 [5,6] or easily produced in industrial plants, such as H2 [7,8] or NH3 [9,10]. Very often transition metal reagents are employed to carry out these processes, with the final aim of developing a true catalytic cycle that directly converts the feedstock species into valuable chemical products.
Another simple, small, molecule that deserves attention is elemental white phosphorus, which is produced in massive amount from natural phosphorite rocks. P4 is indeed readily available and relatively cheap and provides the key for the preparation of the myriad organophosphorus compounds, which are among the chemical specialties with the largest worldwide production. Organophosphorus compounds are synthesized commercially from P4, which, upon oxidation with chlorine to PCl3 or PCl5, is straightforwardly transformed into the target products [11,12]. The reactivity of white phosphorus with transition metal complexes has been extensively investigated in the past few decades and a great variety of transition metal complexes, which contain Pn units originating from either the coupling or degradation of the cage molecule or from the recombination of smaller fragments into polyatomic aggregates, have been described. Such a research field has been exhaustively considered in some reviews [13–17]. Recently, the P4 molecule, which has dangerous environmental effects due to its high reactivity and toxicity, has received specific attention directed both to neutralize the dangerous effects and to achieve its functionalization in mild conditions, that could be of relevance to industrial processes. The former goal has been smartly reached by Nitsche et al. through the reversible incorporation of P4 into an air stable self-assembled tetrahedral capsule [18]. The latter task has been pursued with much effort to achieve the functionalization of white phosphorus in mild conditions. Two approaches have been considered in this respect, either reacting P4 with carbenes and heavier carbene analogues or studying the reactivity of the intact molecule, mediated by the use of transition metal complexes. In the former approach tetraphosphorus frameworks, which contain P atoms with different connectivities and geometries have been obtained [19–27]. Such polyphosphorus units are endowed with high reactivity and, in principle, are useful to obtain organophosphorus derivatives, although up to now such possibilities have been scarcely considered and have not been accomplished yet in a catalytic fashion.
This short review focuses on the second approach, that of the coordination chemistry of white phosphorus and the ensuing activation of the coordinated intact molecule, which has been mostly developed in Florence in the last decade. In the course of these studies several reaction pathways, which are peculiar to coordinated white phosphorus, have been discovered. These have put in evidence the much more controllable reactivity of the P4 tetrahedron once it is coordinated to a transition metal ligand system.
2 Synthesis of intact P4 complexes
The approach to the activation of P4 bound to metal fragments requires the availability in gram amounts of metal complexes containing the intact molecule and then the large-scale synthesis of these generally elusive complexes. Indeed, the number of complexes featuring the intact cage as ligand were limited, up to few years ago, to very few examples, consisting of unstable and/or difficult to handle compounds [28–31]. Recently, 16-electron rhenium [32,33], iron and ruthenium [34] d6-metal platforms have been found useful to straightforwardly coordinate the intact P4 and the related tetraphosphorus trisulfide, P4S3, which presents four phosphorus atoms at the vertices of a tetrahedron and three sulfur atoms in bridging positions [35]. The latter molecule, besides being a possible source of phosphorus derivatives, may be handled with lower risk with respect to P4. In particular, the platforms CpRML2 (CpR = cyclopentadienyl (Cp); pentamethylcyclopentadienyl (Cp*). M = Fe, Ru, Os; L = phosphane donor) form stable and soluble compounds, which may be prepared in gram amounts and may be stored indefinitely at room temperature under nitrogen. The pentamethylcyclopentadienyl derivatives [Cp*ML2(η1-P4)]Y (M = Fe; L2 = dppe. M = Ru; L = PEt3; Y = Cl, BPh4, PF6) are very easily obtained through the spontaneous substitution of P4 for the chloride in the parent [Cp*ML2Cl] [34]. The cyclopentadienyl [CpML2(η1-P4)]Y complexes (M = Ru [36], Os [37]; L = PPh3. M = Fe [38], Ru [39]; L2 = dppe. Y = OTf, PF6) are readily synthesized by removing the coordinated chloride in the starting [CpML2Cl] compound with an appropriate chloride scavenger (AgOTf, TlPF6) in the presence of P4. Almost all the new tetraphosphorus complexes, which are obtained in excellent yields, have been structurally characterized; the crystal structure of the cation [CpRu(PPh3)2(η1-P4)]+ is reported in Fig. 1. The P4 molecule is found to undergo only small deformations upon coordination, which are marginally affected by the nature of the metal and of the coligand(s) and consist in a shortening of the P-P distances formed by the coordinating phosphorus atom with respect to those among the distal uncoordinated P atoms. The cyclopentadienyl ruthenium platform CpRuL2 (L = PPh3 or bidentate phosphine) also coordinates the intact P4S3 cage. However, due to the existence of several distinct potential coordination sites of the molecule, only a mixture of coordination isomers of formula [CpRuL2(η1-P4S3)]PF6 is obtained [35], with the cage bound to the metal either through the apical or one of the three basal P atoms. Compounds with S-coordination were not detected in any of the studied systems. The monometal derivatives, from both cage types, which present phosphorus and sulfur atoms still endowed with coordinating ability, easily react with a second equivalent of the metal synthon CpRuL2 yielding the stable bimetallic [{CpRu(PPh3)2}2(μ,η1:1-P4)](OTf)2 and [{CpRu(PPh3)2}2(μ,η1:1-Papical-Pbasal-P4S3)](OTf)2 compounds, respectively. The crystallographic analyses [40] have shown that the cage molecules tether two ruthenium moieties. A view of the complex bimetallic dication of the P4S3 derivative is reported in Fig. 2. As for the monometallic derivatives, both cages exhibit small distortions from the regular geometry of the free molecule. In particular, in the P4 complex the P–P bond formed by the coordinating phosphorus atoms is the shortest and the opposite bond is the longest among the P–P bonds in the cage. The distortions of the P4S3 cage from regularity essentially involve the two P–S bonds formed by the sulfur atom bound to the coordinating phosphorus atoms, which are slightly shorter (Papical–S) and, respectively, slightly longer (Pbasal–S), than the other P–S bonds in the cage. In order to better investigate the deformations of the cage molecules bound to two different metal platforms, the synthesis of hetero bimetallic complexes has been considered. Only the heterobimetallic compound [{CpRu(PPh3)2}{CpOs(PPh3)2}(μ,η1:1-P4)](OTf)2 has been characterized up to now [37]. Also in such a case the coordinated P4 molecule presents the same deformations observed for the ruthenium homo bimetallic complex, probably due to the similarity of the CpM(PPh3)2 (M = Ru, Os) moieties. 31P NMR data of all compounds clearly show that the cage molecules remain tightly bound to the metal fragments also in solution. The coordinated P4 and P4S3 yield first order spin systems, which are characterized by large downfield shifts of the coordinated phosphorus atoms and a moderate deshielding of the uncoordinated phosphorus atoms with respect to the free molecule values.
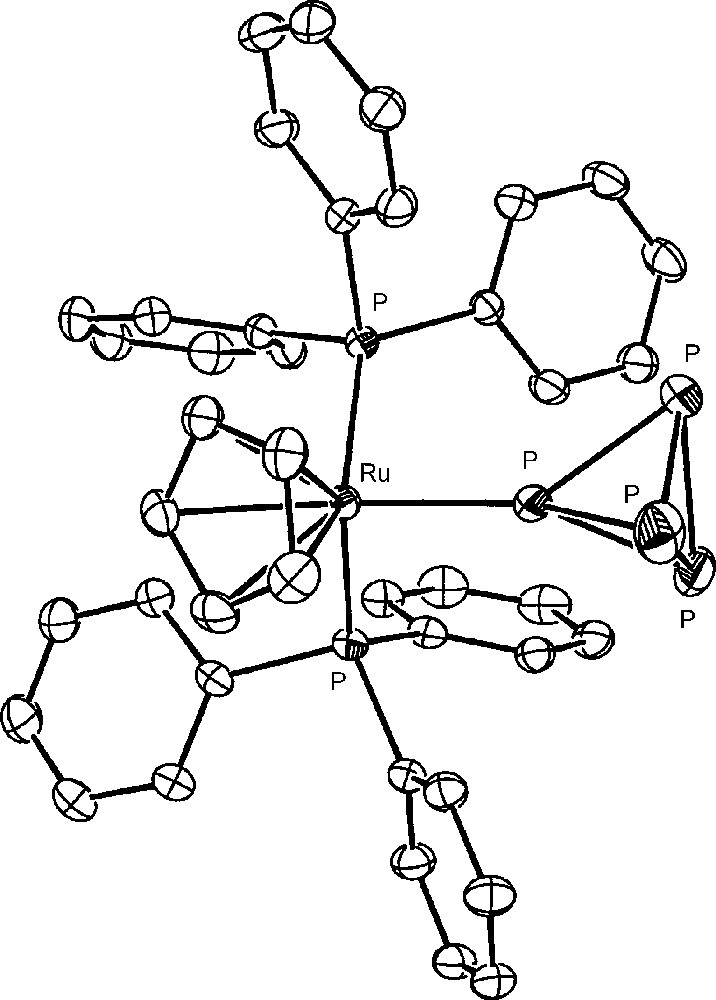
The crystal structure of the cation [CpRu(PPh3)2(η1-P4)]+.
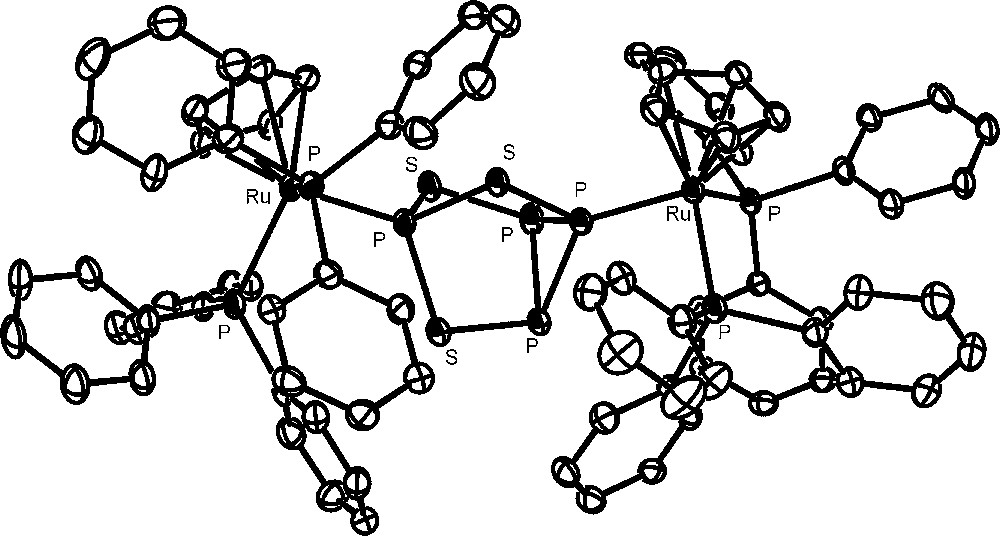
The complex bimetallic dication of the P4S3 derivative.
3 Reactivity of the coordinated P4 and P4S3 cage molecules
The most intriguing property of the coordinated P4 and P4S3 ligands with respect to the free molecules has to do with somewhat spectacular changes of their reactivity. In particular, it is well known from undergraduate chemistry that the cage molecule P4 is stable in the presence of water so that P4 has to be kept and stored under water due to its high reactivity in the presence of air. Also P4S3 does not react with water under ordinary conditions. Coordination of P4, or P4S3, to the metal changes completely this picture and results in a completely different behaviour towards water. Thus, the monometal [CpML2(η1-P4)]Y (L = PPh3; M = Ru [36], Os [37]. L = dppe; M = Ru [39], Fe [38]) derivatives, treated with an excess amount of water, undergo hydrolysis even at room temperature. These complexes yield basically the same hydrolysis products containing oxygenated species, among which hypophosphorous, H3PO2, and phosphorous, H3PO3, acids and PH3, which are trapped through coordination to the CpML2 (M = Ru, Os) platforms in the [CpRuL2(PH3)]Y compounds. The nature of these products reveals that dismutation of the P4 molecule occurs in exceedingly mild conditions, with the coordinated P atom undergoing hydrogenation. Furthermore, the dismutation appears to be regulated by fine electronic and steric properties of the metal fragments as the structurally-related [Cp*Ru(PPh3)2(η1-P4)]Y [34] compound does not undergo hydrolysis in the same conditions. It is known that the catalytic activity of the CpRuL2 moiety (L = monodentate phosphine) is promoted by the dissociation of one phosphine [41]. Such a mechanism, however, should not be available to ruthenium platforms containing bidentate ligands, like dppe, which are reluctant to dissociate from the metal. The identical products obtained from the hydrolysis of the PPh3 and dppe derivatives suggest that the water molecules directly attack the coordinated molecule rather than moving preliminarily into the coordination sphere. In contrast to Ru and Os complexes, the addition of water to the iron [CpFe(dppe)(η1-P4)]Y compound mostly brings about detachment of P4 from the metal [38]; this result, which is consistent with a weaker bond between iron and the P4 molecule, shows that a sufficiently strong interaction between the metal and the cage is required for hydrolysis to occur.
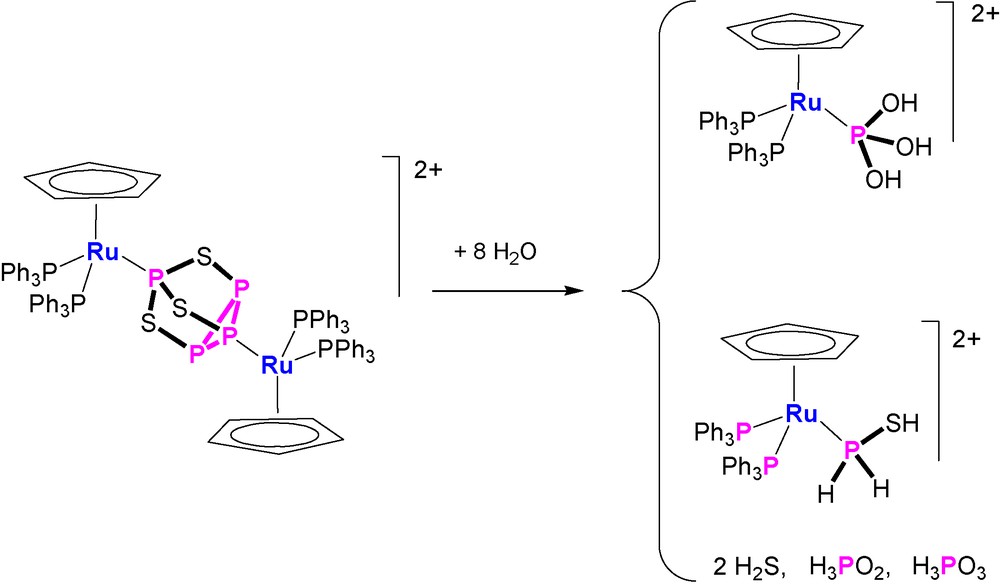
More intriguing and interesting are the reproducible hydrolyses of the bimetallic compounds. The P4S3 derivative [{CpRu(PPh3)2}2(μ,η1:1-Papical-Pbasal-P4S3)](OTf)2 readily reacts at room temperature with an excess amount of water and the outcomings comprise (Scheme 1) hydrogen sulfide, H2S, free hypophosphorous, H3PO2, and phosphorous, H3PO3, acids and the complexes [CpRu(PPh3)2{P(OH)3}]OTf and [CpRu(PPh3)2(PH2SH)]OTf. The four phosphorus products are basically obtained in the same amounts and each one is in a 1:1 ratio with the parent complex. This finding points to the occurrence of a stoichiometric process in which eight molecules of water add regiospecifically to the bridging P4S3 molecule. The [CpRu(PPh3)2{P(OH)3}]OTf complex is an intriguing species as it contains the phosphane tautomer, P(OH)3, of the phosphorous acid bound to the CpRu(PPh3)2 moiety [42]. Remarkably, the pyramidal tautomer, P(OH)3, of the phosphorous acid, which is stabilized by coordination to the ruthenium moiety, is unknown in the free state as the tautomeric equilibrium is almost completely shifted towards the H3PO3 species [42]. In keeping with the formation of such P(OH)3 complex from the controlled hydrolysis of doubly coordinated P4S3, the reaction of [CpRu(PPh3)2Cl] with phosphorous acid results in the quantitative generation of [CpRu(PPh3)2{P(OH)3}]+ via ruthenium promoted tautomerization of H3PO3 into P(OH)3 [42]. The compound [CpRu(PPh3)2(PH2SH)]OTf contains the thiophosphinous acid, PH2SH, coordinated through the phosphorus atom to the CpRu(PPh3)2 platform, Fig. 3. The stability of the coordinated PH2SH molecule, which furthermore may be easily deprotonated and alkylated [43], is quite remarkable in view the extreme instability of the free compound that has been obtained in very harsh conditions [44,45]. By considering the oxidation numbers of the P4S3 atoms and those of the outcomings, it appears that the addition of water promotes dismutation of only two P atoms of the coordinated cage. In fact, the sulphur atoms of P4S3, yielding H2S−II and PH2S−IIH, do not change their oxidation number during the hydrolysis. The apical and the three basal phosphorus atoms of the free cage have oxidation numbers +III and +I, respectively, and they yield H3P+IO2, P−IH2SH and two molecules of H3P+IIIO3. According to these experimental findings and assuming that the coordinated phosphorus atoms undergo their transformations in the metal coordination sphere, it is likely that the complex [CpRu(PPh3)2{P(OH)3}]OTf forms from the hydroxylation of the apical phosphorus, which therefore does not change its oxidation state. In this mechanistic picture, the thiophosphinous acid derivative, [CpRu(PPh3)2(PH2SH)]OTf, is likely obtained from the reduction of the coordinated basal phosphorus, that formally acquires two electrons from one of the uncoordinated P-atoms. The latter P atoms, after adding water, form H3P+IIIO3 and H3P+IO2, which are among the observed products in a 1:1 ratio with respect to each other and to the metal coordinated P(OH)3 and PH2SH molecules.
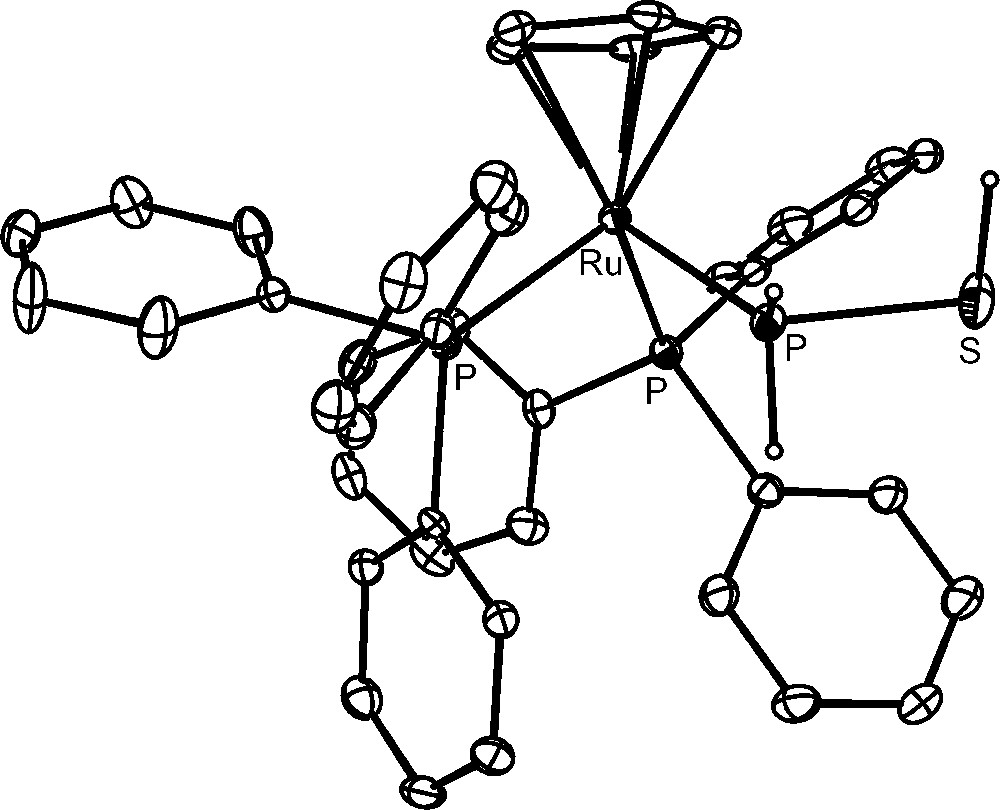
The compound [CpRu(PPh3)2(PH2SH)]OTf.
The hydrolysis of the bimetallic P4 derivative [{CpRu(PPh3)2}2(μ,η1:1-P4)](OTf)2 is even more intriguing for both the nature of the products and the time needed to complete the reaction are strongly affected by the relative amounts of the complex and water. The dinuclear complex is completely hydrolyzed within 6 days in the presence of 20 equivalents of water to yield a mixture of several products. The two most abundant derivatives, ca. 65% of the parent compound, are [{CpRu(PPh3)2}{CpRu(PPh3)}{μ1,4:3,η2:1-P(OH)2PHPHPH(OH)}](OTf)2 and [{CpRu(PPh3)2}{CpRu(PPh3)}{P(OH)3)}(μ,η1:1-P2H4)](OTf)2. The remaining species comprise the new [{CpRu(PPh3)2}2(μ,η1:1-P2H4)](OTf)2 and the known [CpRu(PPh3)2{PR(OH)2}]OTf (R = H, OH) and [CpRu(PPh3)2(PH3)]OTf compounds, as well as free phosphorous acid, H3PO3. The crystal structure of the first of these complexes, Fig. 4, presents the 1,1,4-tris-hydroxytetraphosphane molecule, P(OH)2PHPHPH(OH), which chelates with its two hydroxylated ends the metal atom bearing one triphenylphosphane and is bound through one of its intermediate phosphorus atoms to the other metal atom in the cation, which is still coordinated by both triphenylphosphanes, as in the original {CpRu(PPh3)2} fragment. Notably, the complex spin system of the 31P NMR spectrum of the compound is in line with that expected on the basis of the X-ray structure, pointing to a good stability of the coordinated molecule also in solution. The 1,1,4-tris-hydroxytetraphosphane molecule, stabilised by μ,η2:1-coordination to two {CpRu} moieties, is particularly intriguing as it represents a member of the practically unknown family of polyhydroxyphosphanes, i.e., compounds of general formula PnH[(n+2)−m](OH)m (m ≤ n + 2), which may be related to the slightly more investigated, but still elusive, family of polyphosphorus hydrides (PnHn+2), by substitution of OH groups for one or more H atoms of the hydrides. The latter linear phosphanes are formally isoelectronic with the corresponding alkanes but are quite reactive, undergoing exothermic decomposition through facile disproportionation, even at low temperature [46]. Remarkably, no compound formed by their reaction with oxygen has been fully characterized, as they spontaneously ignite yielding, as the final product, phosphoric acid.
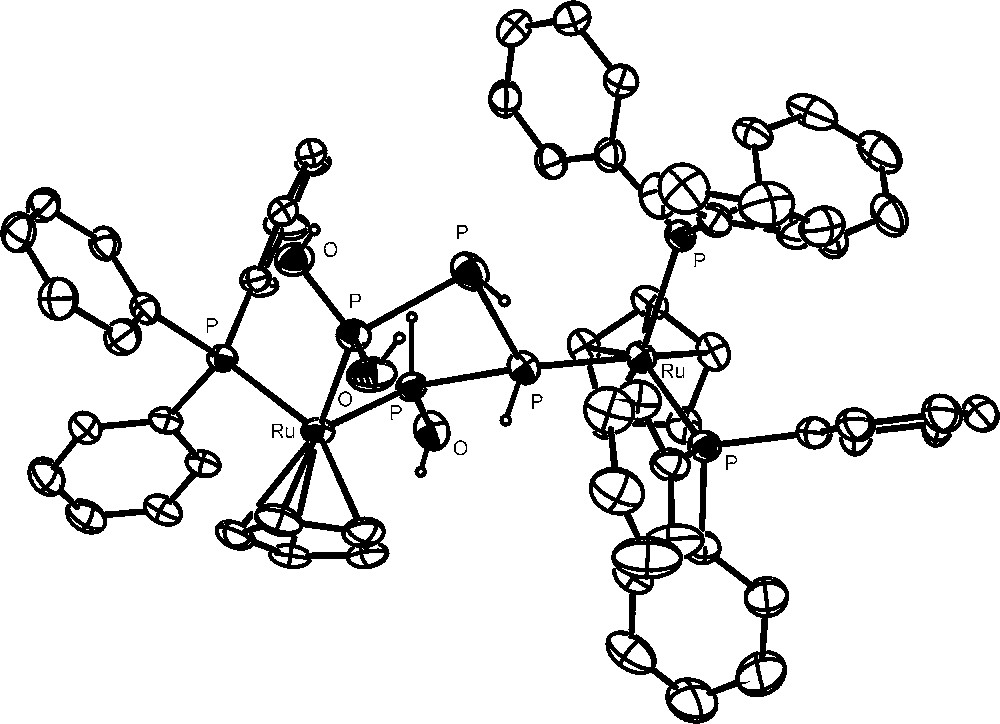
The 1,1,4-tris-hydroxytetraphosphane molecule, P(OH)2PHPHPH(OH).
The complexes [{CpRu(PPh3)2}2(μ,η1:1-P2H4)](OTf)2 (crystal structure of the cation is shown in Fig. 5) and [{CpRu(PPh3)2}{CpRu(PPh3){P(OH)3)}(μ,η1:1-P2H4)](OTf)2 contain the diphosphane, P2H4, molecule bridging two ruthenium moieties: these are equivalent in the former complex, whereas in the latter one of them has likely undergone substitution of a molecule of phosphorous acid for one of the triphenylphosphanes. These compounds provide the first fully characterized examples of complexes containing the highly reactive P2H4 ligand. Remarkably, the diphosphane molecule, which in the free state can be handled only under special safety precautions, is incredibly stabilized through coordination to two ruthenium moieties [46].
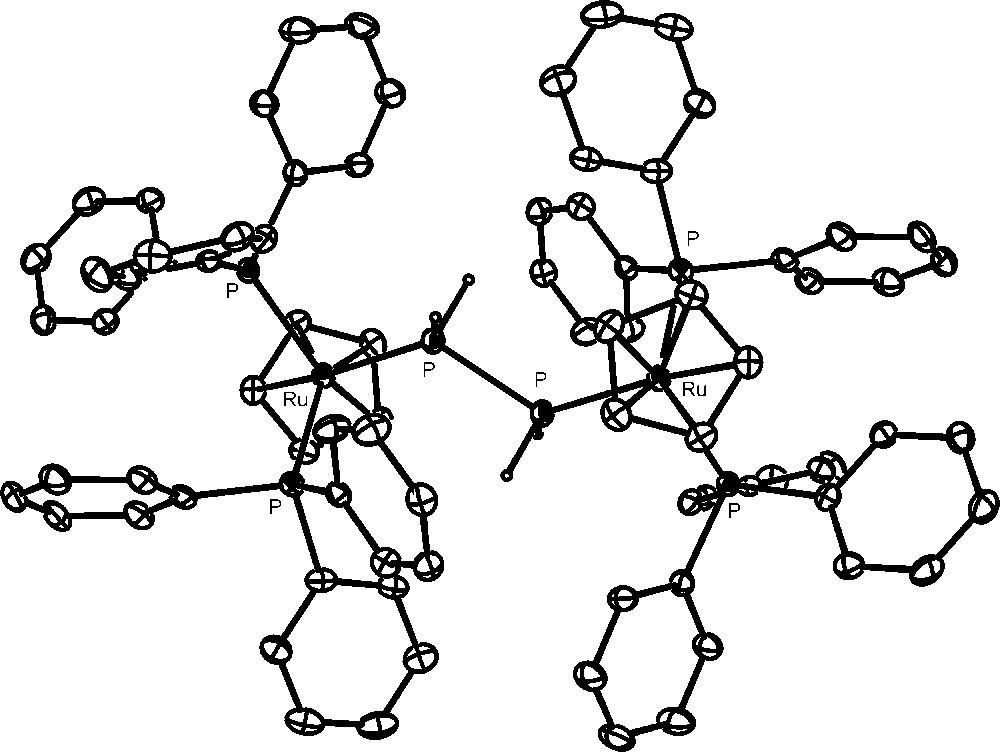
Crystal structure of the cation, [{CpRu(PPh3)2}2(μ,η1:1-P2H4)](OTf)2.
The hydrolysis in the presence of a larger amount of water (1:100) reaches completion in a few hours and the outcomings contain free phosphorous acid and the compounds, [{CpRu(PPh3)2}2(P2H4)](OTf)2, [CpRu(PPh3)2{PR(OH)2}](OTf) (R = H, OH) and [CpRu(PPh3)2(PH3)](OTf), which have been described above. It is worthwhile to note that such reaction conditions favour the formation of phosphorus compounds that contain only one or two phosphorus atoms from the starting P4 via further hydrolytic degradation of intermediate polyhydroxylated polyphosphorus chains.
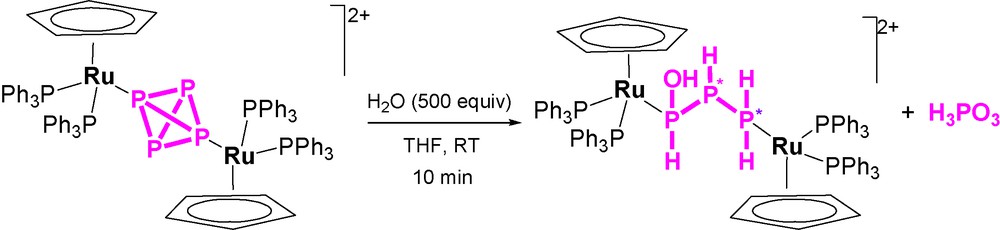
Unexpectedly, the addition of 500 equivalents of water to one equivalent of the complex yields completion of the hydrolysis in a few minutes through a simple process (Scheme 2) that leads to the formation of one equivalent of H3PO3 and to the new complex [{CpRu(PPh3)2}2{μ1:3,η1:1-PH(OH)PHPH2}](OTf)2 (the crystal structure of the cation is shown in Fig. 6), which contains the previously unknown 1-hydroxytriphosphane, PH(OH)PHPH2 bridging two CpRu(PPh3)2 moieties through the phosphorus atoms of the PH(OH) and PH2 end groups. The NMR data confirm that the solid-state structure is retained in solution and substantiates the presence of two diastereoisomers in agreement with the presence of two P-chiral atoms in the coordinated triphosphane. Such outcomings point to the occurrence of a stoichiometric process in which four molecules of water formally add to the bridging P4 ligand. The addition favours a disproportionative redox process where three electrons are removed from one P atom of the tetraphosphorus, which is consequently oxidized to H3PO3. These three electrons are unequally redristibuted among the remaining three P atoms of the P4 molecule in the simultaneous reductive process of the disproportionation to yield the 1-hydroxytriphosphane, P0H(OH)P−IHP−IIH2, which is eventually stabilized by coordination to the ruthenium atoms.
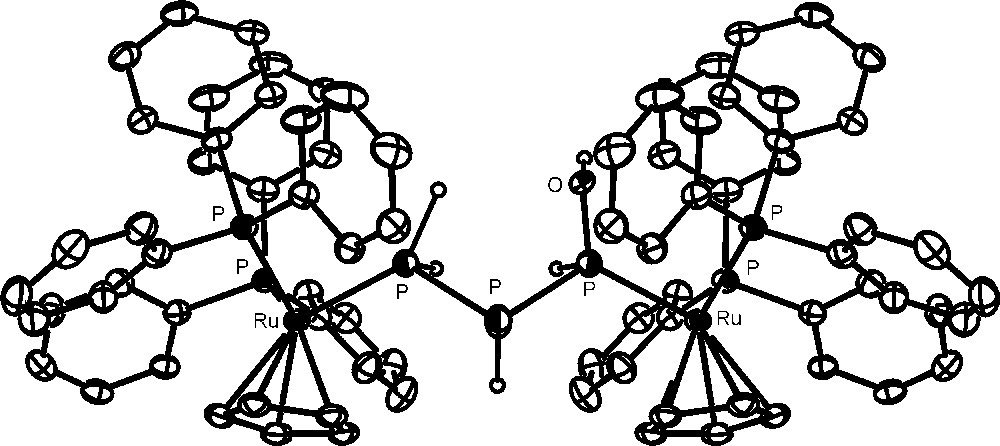
Crystal structure of the cation, [{CpRu(PPh3)2}2{μ1:3,η1:1-PH(OH)PHPH2}](OTf)2.
The numerous species containing up to four phosphorus atoms, i.e. PH3, PH(OH)2, P(OH)3, P2H4, PH2PHPH(OH) and P(OH)2PHPHPH(OH), obtained by the various processes of hydrolysis may be assumed as thermodynamic minima along the degradation pathway of the dimetallated P4 ligand. Their number and nature suggest that several routes may be activated during the hydrolysis, depending on the reaction conditions, particularly on the amount of added water. Therefore, although no rigorous and comprehensive mechanistic picture of the hydrolysis of the dimetallated P4 may be proposed in the absence of an in-depth kinetic study, it nevertheless seems to be possible to trace some hypotheses about the initial steps of the process, particularly those leading to formation of 1,1,4-tris-(hydroxy)tetraphosphane. Indeed the formation of the P(OH)2PHPHPH(OH) suggests that these species may result from the addition of three water molecules to the doubly coordinated P4. An assignment of oxidation numbers shows that a disproportionative reshuffle occurs, yielding P-atoms encompassing the oxidation numbers +2, −1, −1 and 0 for the four P atoms of the molecule, in the above order. A pictorial description of a possible route for the stepwise addition of three water molecules to 1, with cleavage of three P–P bonds, however, without total disruption of the diruthenium–tetraphosphorus assembly, is shown in Scheme 3. Whatever the detailed order of addition may be, one may assume that the first two water molecules add across the P–P bonds connecting the pair of unmetallated P atoms and across the opposite bond connecting the metallated pair, yielding the (undetected) species 1,2-(dihydroxy)tetraphosphacyclobutane (A) 1,3-dicoordinated to two {CpRu(PPh3)2} moieties. Then, in order that formation of the 1,1,4-tris(hydroxy)tetraphosphane is rationalised, the addition of the third water molecule to A should take place with complete 1,2-regioselectivity, resulting in the intermediate B. From this point on, the process should be straightforward and formation of [{CpRu(PPh3)2}{CpRu(PPh3)}{μ1,4:3,η2:1-P(OH)2PHPHPH(OH)}](OTf)2 would be brought about by coordination of the pending PH(OH) end of the opened tetraphosphane to the ruthenium center already coordinated by the distal P(OH)2 end, with associated decoordination of one triphenylphosphane ligand. By contrast, the alternative addition of the third water molecule across the 3,4-P–P bond in A would yield, after passing through C and the addition of a fourth water molecule, the triphosphorus chain featuring the 1-hydroxytriphosphane ligand, PH(OH)PHPH2, which has been characterized in the complex [{CpRu(PPh3)2}2{μ1:3,η1:1-PH2PHPH(OH)}](OTf)2. The formation of this compound is accompanied by that of one equivalent of P(OH)3, which, once released in the reaction medium, retrotautomerises to the more stable oxyacid form, H3PO3.
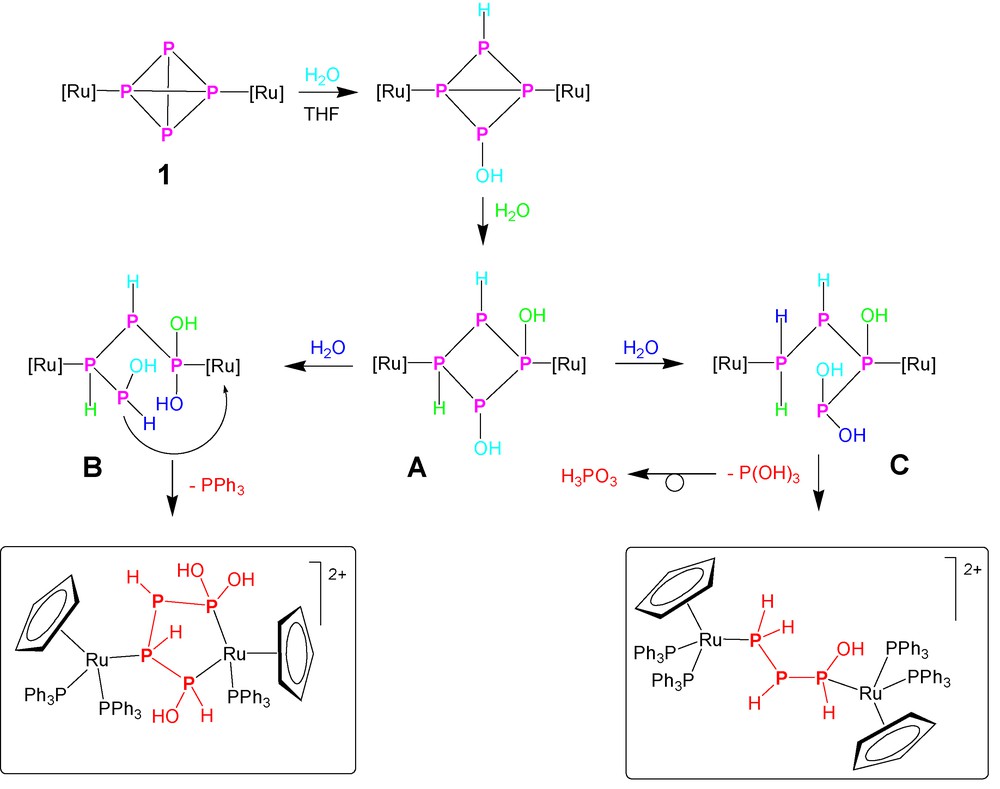
From this point on the mechanism of the hydrolytic reaction should be unambiguous asking for the addition of two further molecules of water to go to completion. This last hydrolytic step results in the intermediate formation of the diphosphane compound [{CpRu(PPh3)2}2(P2H4)](OTf)2, and, at the very end of the process, the two monophosphorus compounds [CpRu(PPh3)2{PH2(OH)}](OTf) and [CpRu(PPh3)2(PH3)](OTf) should be formed. Noticeably, while the PH3 species has been isolated and thoroughly characterized [36,37], we have not yet been able to detect the formation of PH2OH either as a coordinated ligand or a free molecule. The formation of the elusive phosphinous acid, PH2OH, molecule, which represents the phosphane tautomer of the unknown PH3 oxide, finds some justification and mechanistic support in the formation of the related PH2SH molecule, which has been detected and crystallographycally characterised in the ruthenium complex [CpRu(PPh3)2{PH2(SH)}](OTf) obtained from the hydrolysis of the P4S3 derivative [40].
Acknowledgement
Financial support by the Italian Ministero dell’Istruzione, dell’Università e della Ricerca is gratefully acknowledged (PRIN2007). The work has been carried out in the frame of COST ACTION CM0802 “PHOSCINET” of the European Community.