

1 Introduction
Carbon dioxide utilization as chemical feedstock could have currently a significant role in addressing the emission excess of CO2 in the atmosphere, but more importantly transforming an end-product into a valuable feedstock [1]. Carbon dioxide has been employed for decades as a raw material for the production of urea, salicylic acid, and ethylene (propylene) carbonates, as well as for methanol synthesis from a blend of CO and CO2. Recent interest in the use of CO2 as feedstock is motivated by the development of new reactions from so-called renewable carbon. However, CO2 displays a limited reactivity when compared to other C1 molecules such as carbon monoxide and phosgene. Nevertheless, the need for friendly reactants, products, and safer processes according to the principles of Green Chemistry and Engineering [2] offers the opportunity to investigate new reaction pathways based on CO2.
The alternative to phosgene is a challenging option that may open new perspectives for carbonate and carbamate synthesis including polymers [3]. The phosgene technology has still a leading role in a range of industries, including production of pharmaceuticals, agrochemicals, and others that raises obvious technical and economical barriers. Phosgene is widely recognized as one of the most acutely toxic substances used in commerce today. It is currently produced from brine and CO in a two-step process: electrolysis of brine for forming Cl2 (chlor-alkali process) which is further reacted with CO on activated charcoal catalyst leading to COCl2 [4]. In the case study of organic carbonate synthesis, the reaction co-produces HCl. Thus, the chlorine cycle Cl- → Cl2 → Cl- is, overall, energy-intensive. Phosgene-free production of DMC is mainly based on the oxidative carbonylation of methanol which limits scale-up due to inherent hazards [5]. Nevertheless, much attention is being focused on extending DMC uses as intermediate, e.g. in organic synthesis [6], phosgene-free synthesis of polymers [7], and gasoline blending [8]. As a matter of fact, DMC fulfils green chemistry criteria [2]. It has a low toxicity, an absence of any irritant or mutagenic effects and low atmospheric loss, and is highly biodegradable [7,9]. Therefore, there is a real need for friendly synthetic routes to DMC.
Table 1 reports the various alternative feedstocks of current interest to get DMC. The atom economy factor (AE) is also given as a helpful indicator of the amount of co-produced waste to assess the greenness of a reaction [10].
The different feedstocks to DMC, and the corresponding atom economy factor (AE).
| Entry | Feedstock | Product | AE (wt%) |
| 1 | COCl2 + 2 CH3OH | DMC + 2 HCl | 55 |
| 2 | CO + 1/2 O2 + 2 CH3OH | DMC + H2O | 83 |
| 3 | (CH2O)2CO + 2 CH3OH | DMC + HO(CH2)2OH | 59 |
| 4 | (NH3)2CO + 2 CH3OH | DMC + 2 NH3 | 72 |
| 5 | CO2 + 2 CH3OH | DMC + H2O | 83 |
The conventional phosgene route (entry 1) gives an AE factor of 55 wt% which shows that nearly half the weight of the reactants is transformed into hydrogen chloride. A much more favorable factor (83 wt%) is obtained from the on-stream oxidative carbonylation of methanol with the co-formation of water (entry 2). Entry 3 corresponds to the transesterification of ethylene carbonate which co-produces ethylene glycol leading to a decrease of the AE value to 59 wt%. The methanolysis of urea is more favorable (AE = 72 wt%). Moreover, the co-formed ammonia may be recycled for urea synthesis. It is worthy of note that entries 3 and 4 correspond to an indirect use of CO2 since ethylene carbonate and urea are thereof industrially produced. Entry 5 highlights that the direct synthesis of DMC from CO2 and methanol leads, as entry 2, to the highest AE factor of 83 wt%. Interestingly, in the context of non-fossil carbon resources for making organics coupling methanol with CO2 presents the unique advantage of utilizing CO2 as carbon feedstock, provided that methanol is thereof obtained by hydrogenation [11].
The key role played by DMC and CO2 in the practice of green chemistry has stimulated fundamental research for developing innovative catalytic methodologies. Progress in this field has been slow, however, mainly due to the low reactivity of CO2. The current main issue for the reaction reported in entry 5 of Table 1 is to enhance the yield and rate for DMC formation. One option consists in adding water traps to the catalytic systems for preventing catalyst deactivation by water as well as to shifting the equilibrium [12]. Nevertheless, there is a need for exploring new catalysts for rate enhancement in the absence of water trap additives. In recent years, a variety of heterogeneous catalysts has been screened. The reaction is commonly run under high pressure, i.e. 100–300 bar, at 130–180 °C over ZrO2-based catalysts [13], CeO2 [14], Cu-Ni/graphite [15], V-doped Cu–Ni/AC catalysts [16], and polyoxometalates [17]. Zirconia-based catalysts are those exhibiting the highest selectivity to DMC (∼100%). Interestingly, organometallic complexes such as diorganotin (IV) compounds are also active precursors under the same reaction conditions. Importantly enough, they are superior to heterogeneous catalysts in terms of atom economy since they are totally selective [12(b),18]. However, organometallic tin compounds in general suffer from environmental pressure due to issues around tributyltin derivatives.
As part of our ongoing research on tin compounds for the synthesis of DMC from methanol and CO2, we herein report preliminary results on the catalytic behavior of supported SnO2 catalysts for comparison with unsupported SnO2 and diorganotin (IV) compounds. The synthesis of unsupported SnO2 was achieved by a free-chloride sol–gel route from tin tetra-tert-butoxide precursor providing SnO2 nanoparticles with an average diameter of 4 nm [19]. Supported SnO2 was prepared from the same tin precursor with the objective to avoid handling nanoparticles. A preliminary screening with alumina, silica-alumina, and silica as supports showed that silica was the best candidate due to its inertness towards methanol dehydration and DMC decomposition. ZrO2 catalysts were also prepared following identical methods to get a reference under our reaction conditions since they are known to be the most selective to DMC among the heterogeneous catalysts reported so far [13]. The synthesis of the silica-supported tin and zirconium oxides is also reported as there is no previous report.
2 Results and discussion
2.1 Preparation of silica-supported tin and zirconium oxides from Sn(OtBu)4 and Zr(OtBu)4
Supported catalysts may exhibit different structure-activity relationship depending on the dispersion of the active phase, metal loading, and interaction with the support. In most cases, with catalysts prepared by a conventional impregnation method from inorganic salts, dispersion of the active phase is difficult [20]. Using metalorganic compounds as precursors allows a better control of this dispersion [21]. Accordingly, monomeric metal alkoxides were chosen in this work as convenient precursors for reacting with the hydroxyl groups of the support, thus allowing chemical grafting. Tin and zirconium tetra-tert-alkoxide dissolved in CH2Cl2 were contacted, respectively, with silica (Aerosil Degussa 200, SSA = 196 m2 g−1) pretreated at 450 °C. The initial M/SiO2 (M = Sn, Zr) ratio was varied from 10 to 20 wt%. After filtration, washings with CH2Cl2 and drying at 40 °C under vacuum, the solids contained 4.9 and 4.4 ± 0.3 wt% of tin and zirconium, respectively. These metal contents remain unchanged when the reaction is performed either at 25 or 60 °C. Further washings with tetrahydrofuran or methanol did not promote metal leaching. These results show that chemical grafting of the complexes did occur at the saturation value of 1.3 ± 0.1 Sn (or Zr) per nm2. As the OH content of silica pretreated at 450 °C is about 2 OH per nm2 [22], they may have reacted through two of the four tert-butoxy ligands. However, using other monomers such as nBu3SnOCH3 and nBu2Sn(OtBu)2 has no noticable changes in the loading value. As it is known that only the methoxy ligand of nBu3SnOCH3 undergoes protolysis [23], it can, therefore, be inferred that the saturation loading under these experimental conditions is mainly controlled by steric effects.
The thermal stability of the anchored tert-butoxy tin species was studied and first evaluated by the temperature-programmed desorption technique (TPD) using a thermal conductivity detector (TCD). As shown in Fig. 1, the onset of decomposition occurs at 100 °C over a narrow range of temperature with a major peak at 180 °C. Analysis of the gas evolved by gas chromatography (GC) revealed the main presence of isobutene.
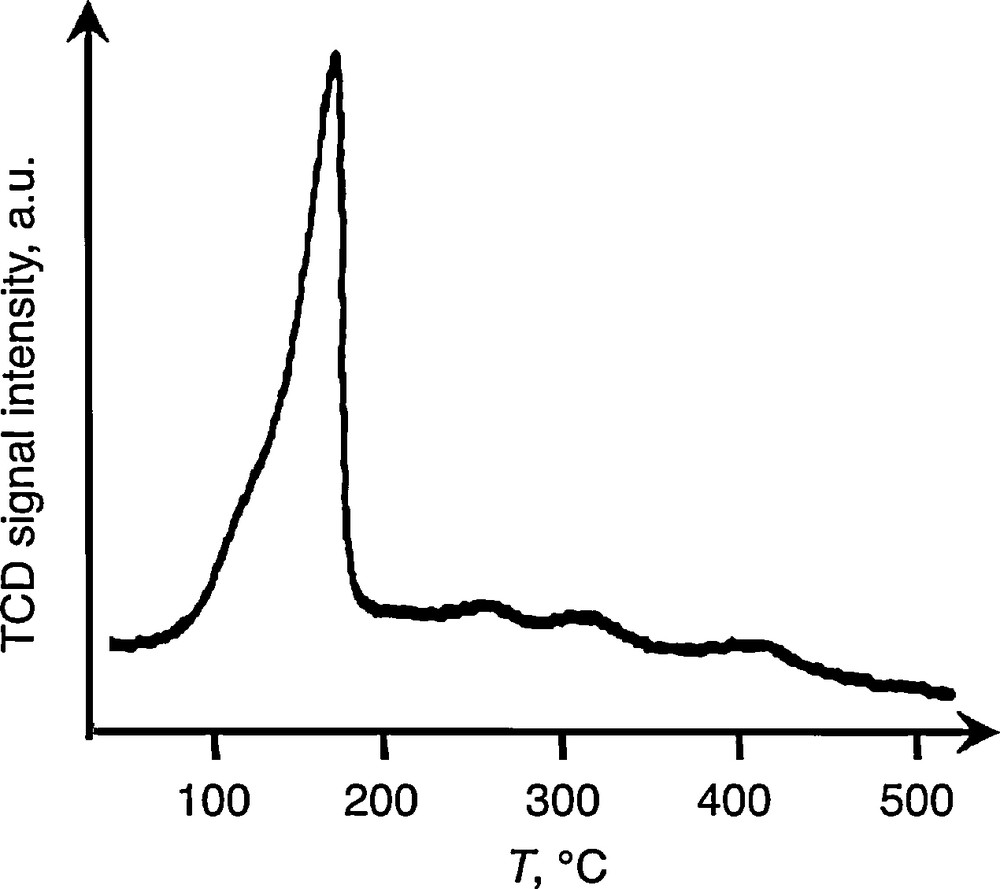
TPD profile under helium of Sn(OtBu)4 adsorbed on SiO2 (4.9 Sn wt%).
Investigation of the grafting mode was performed by FTIR transmission spectroscopy (Fig. 2). The IR spectrum of pure silica pretreated at 450 °C shows the presence of a sharp ν(OH) band at 3747 cm−1 characteristic of isolated silanol groups and a weak shoulder at ∼3675 cm−1 due to the presence of H-bonded silanols (Fig. 2a) in agreement with the literature [24].
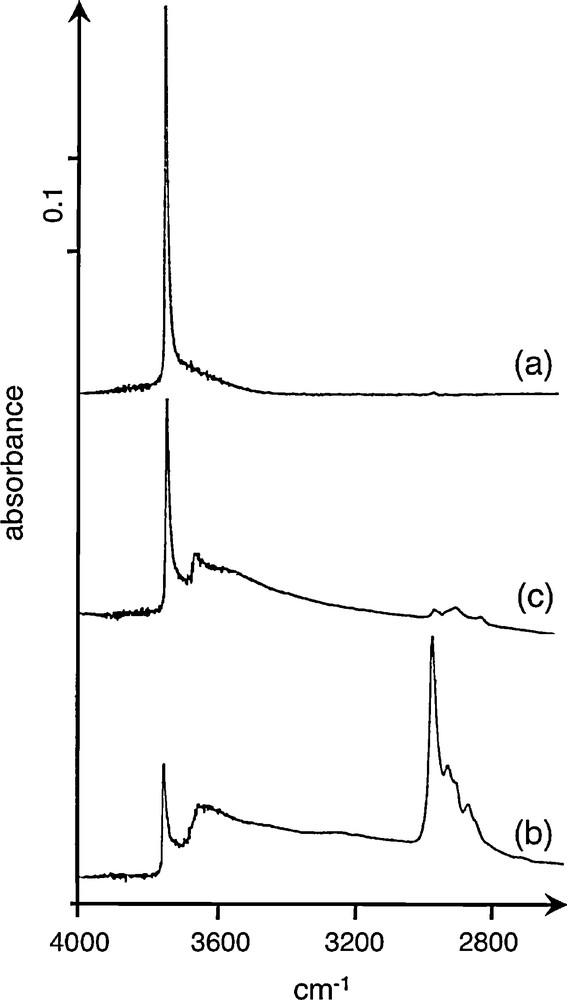
IR spectra in the 4000-2600 cm-l region: (a) SiO2 pretreated at 450 °C, (b) adsorption of Sn(OtBu)4 (5 Sn wt%) at 25 °C followed by vacuum for 4 h, (c) heat-treatment at 110 °C for 4 h followed by vacuum for 4 h.
Adsorption of Sn(OtBu)4 at room temperature led to the consumption of isolated silanol groups, leaving a broad band at ca. 3620 cm−1 (Fig. 2b). After heating to 110 °C followed by evacuation, the ν(CH) bands of the tert-butyl groups have almost disappeared while the ν(OH) band of the isolated silanols is partially restored (Fig. 2c). Although it was not our objective to study the reaction mechanism involved, the restoration of SiOH groups and the formation of isobutene analyzed as analyzed by TPD experiments in the same temperature range are likely due to a dehydration process. Such a reaction is induced by protolysis of tin tert-butoxide by the isolated OH groups. These results show that the extrusion of the tert-butoxy ligands is occurring under mild conditions restoring partially the hydroxyl groups of silica. Further work is presently under study to check whether silica-supported zirconium tert-butoxide behaves in a similar manner. After a vacuum treatment at 130 °C, the BET surface area of silica slightly decreased with supported tin from 196 to 181 m2 g−1, and with supported zirconium to 186 m2 g−1.
2.2 Dimethyl carbonate synthesis from methanol and CO2 on unsupported ZrO2 and SnO2 and silica-supported catalysts
The direct synthesis of dimethyl carbonate from methanol and CO2 is usually conducted in the temperature range 120–180 °C. The reaction takes place under CO2 pressure due to the reversibility of carbon dioxide insertion into MOCH3 moieties leading to the methyl carbonato species, MOCO2CH3, a key species in the reaction mechanism [18(b),25]. The catalytic experiments herein described were performed at 150 °C under 200 bar in a batch autoclave. Under these conditions, the reactants are forming a single dense phase [26].
Our previous study with unsupported SnO2 and ZrO2 catalysts showed that the DMC yield linearly increased with the specific surface area (SSA) of the oxides [19]. High SSA up to 170 m2 g−1 could be obtained by a sol–gel procedure from tin- and zirconium tetra-tert-butoxide thanks to the formation of oxide grains in the nanometric range (4 nm in average). Fig. 3 reports the activity for DMC synthesis expressed as the DMC/M (M= Zr, Sn) molar ratio for ZrO2 and SnO2 samples with SSA of 172 and 170 m2 g−1, respectively. In the case of ZrO2 (Fig. 3a), the DMC yield increases steadily for several hours of reaction before reaching a plateau. The reaction was quite selective in DMC. During the first 6 h of a run, no other organics could be detected. However, dimethyl ether (DME) was found in trace amounts at longer reaction times. Its formation is likely due to methanol dehydration. The co-produced water may inhibit DMC formation under batch conditions as it accumulates in the autoclave which might explain the DMC plateau after 80 h run.
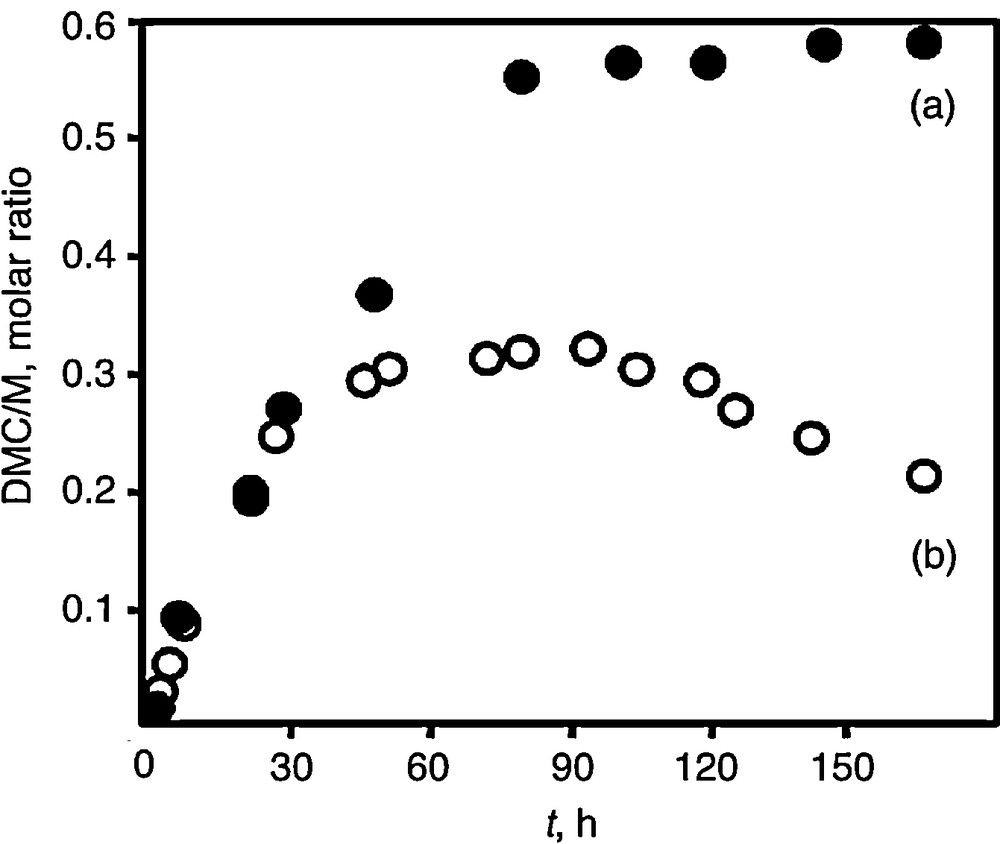
DMC/M molar ratio (M = Sn or Zr) as a function of reaction time at 150 °C and 200 bar: (a) ZrO2 (172 m2 g−1) and (b) SnO2 (170 m2 g−1).
Interestingly, the SnO2 sample exhibits a similar activity to DMC at the beginning of the run. Nevertheless, a noticeable decrease in DMC yield beyond the 100 h run is observed (Fig. 3b). As a matter of fact, the reaction was found to be non-selective in DMC. Almost equal amounts of DME were analyzed. Therefore, the additional accumulation of water in the reactor likely contributes to DMC decomposition with prolonged reaction time. Indeed, the activity was restored after ex-situ reactivation of the catalysts at 130 °C for 2 h under vacuum. After four recyclings of ZrO2, the DMC yield remained identical. The same observation stands for SnO2 with which extensive recyclings were performed after reaction times ranging from 12 to 60 h. With cumulative reaction duration of 17 days with SnO2, the SSA was slightly decreased by 14%. Thus, it can be concluded that the catalysts exhibit excellent stability at the lab scale.
Our next objective was to compare under the same experimental conditions the activity of silica-supported SnO2 and ZrO2 prepared from tin- and zirconium tetrabutoxide, thus avoiding the issue of handling nanoparticles. The metal loading was fixed at the saturation limit as determined by the aforementioned grafting study, but the impregnation method of the metal tetrabutoxide was applied. As heating to 130 °C under vacuum induces the elimination of the butoxy ligands (see precedent section), the supported samples were pretreated under this condition giving a final tin and zirconium loading of 4.9 and 3.1 wt%, respectively. The corresponding catalysts were tested for DMC synthesis.
Fig. 4 reports the DMC yield expressed as the DMC/M (M= Zr, Sn) molar ratio with time for comparison with the unsupported catalysts. As observed in Fig. 3 for the unsupported oxides, the DMC yield increases steadily with reaction time over several hours.
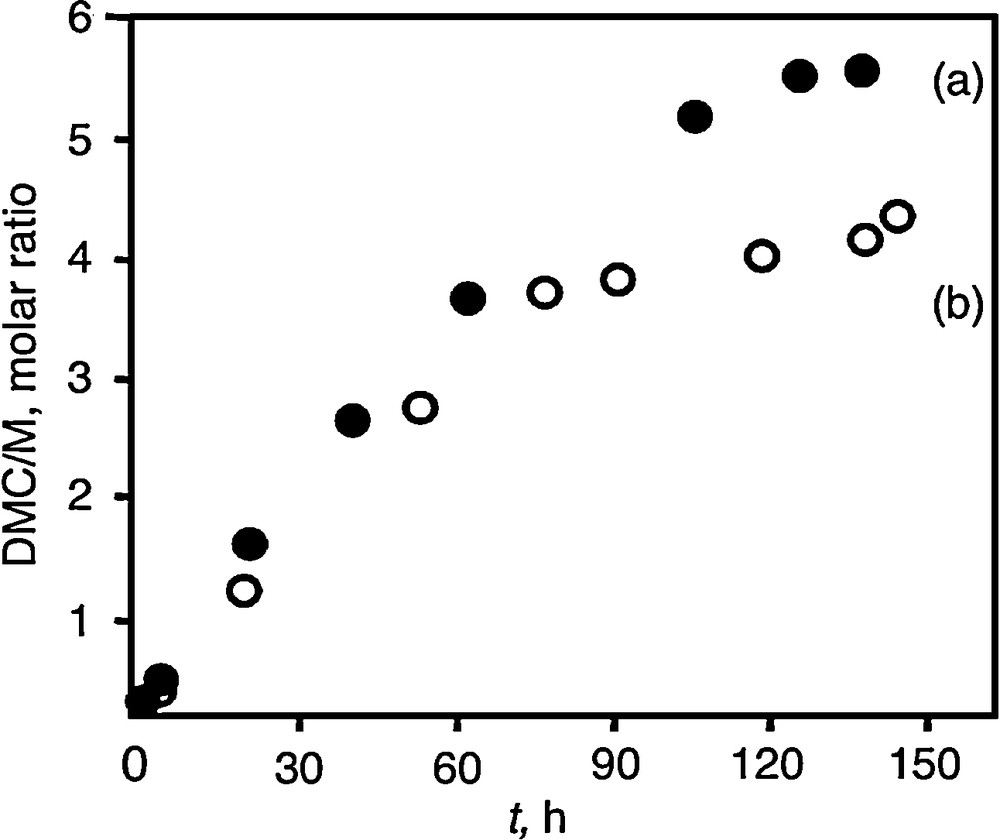
DMC/M molar ratio (M = Sn or Zr) as a function of reaction time at 150 °C and 200 bar: (a) ZrO2/SiO2 (3.1 Zr wt%), (b) SnO2/SiO2 (4.9 Sn wt%).
With ZrO2/SiO2, a plateau is again observed but for higher reaction time than for unsupported ZrO2, i.e. 120 vs 70 h (Fig. 4a). Supported SnO2 exhibits a similar activity compared to supported ZrO2 during the first 80 h, a reaction time also higher than for unsupported SnO2 (Fig. 4b). Then, the rate of DMC formation is somewhat decreased. DME was detected but not analyzed quantitatively. This will be done in the near future for assessing the selectivity. However, it is likely that the formation of water during DME synthesis hinders the reaction to DMC as for unsupported SnO2. Indeed, the activity was restored after ex-situ reactivation of the catalysts at 130 °C for 2 h under vacuum.
At the beginning of the runs, the rate of DMC formation is nearly identical on tin- and zirconium-supported catalysts. Importantly enough, there is a drastic enhancement in comparison with unsupported catalysts. The observed difference is of an order of magnitude which demonstrates the superiority of the supported catalysts. Although it is premature to draw any correlation between structure and activity to rationalize these high turnover numbers, the physicochemical properties of the supported active species at low coverage may be influenced by the support interaction. Further work is in progress for characterization and optimization of the activity with metal loading. The results of DMC formation on silica-supported tin oxide can be compared with those we previously obtained with the homogeneous nBu2Sn(OCH3)2 and tertBu2Sn(OCH3)2 precursors under the same experimental conditions [26,27]. The organometallics are totally selective to DMC whereas tin oxide also produces DME. The initial rate of DMC formation during the first 6 h of the run is about twice with the organometallics. However, they fastly desactivate due to intermolecular rearrangement that leads to polynuclear tin species. The SnO2/SiO2 and ZrO2/SiO2 systems are much more stable providing an almost constant rate for DMC over 60 h. The consequence is much higher turnover numbers.
3 Conclusion
We have shown that tin and zirconium oxides catalyze the reaction of CO2 with methanol to afford DMC. Using silica-supported catalysts, it was possible to get a stable activity over several hours at 150 °C and 200 bar pressure as well as to achieve many recyclings without loss of activity. The observed selectivity for DMC was much higher with unsupported zirconium oxide and will be assessed quantitatively on silica-supported catalysts for comparison. However, there is almost an order of magnitude in the turnover numbers to DMC with the supported catalysts. The turnover numbers are also much higher than with the organotins we studied but selectivity and initial rate are lower. Silica support likely contributes to promote and stabilize the active species. Better dispersion and/or specific interaction may be at the origin of this effect. Overall, this work highlights for the first time that supported tin- and zirconium oxides may compete with soluble organotin precursors with a definite advantage for the greenness of the catalysts.
4 Experimental
All the syntheses were performed under argon atmosphere using the Schlenk tube technique. The solvents (purum grade) were deoxygenated, dried using standard techniques, then stored under argon. Tin tetra-tert-butoxide was prepared according to ref. [28] and zirconium tetra-tert-butoxide was purchased from Aldrich. Silica (Degussa Aerosil 200, SSA = 196 m2 g−1) was pretreated under vacuum at 450 °C for 12 h prior the reaction with the metal alkoxides.
4.1 BET surface area
Nitrogen adsorption data were obtained at –196 °C with a Micromeritics ASAP 2020 apparatus. The specific surface areas (SSA) were calculated from the BET method.
4.2 Transmission FTIR
The spectra were recorded at room temperature on a FT 1725X Perkin-Elmer spectrometer (2 cm-1 resolution) equipped with IRDM software. A self-supported disk of silica (∼15 mg) was placed in a quartz cell equipped with CaF2 windows, then evacuated to 450 °C for 12 h prior to Sn(OtBu)4 adsorption. A CH2Cl2 solution of Sn(OtBu)4 was deposited dropwise onto the silica disk under argon in a glove box, in order to get around 5 wt% of metal loading. Then, the solvent was evacuated under vacuum at room temperature for 4 h.
4.3 Temperature programmed desorption (TPD)
TPD experiments were conducted under a helium flow rate of 10 cm3 min−1 in a microreactor with 76 mg of sample at a heating rate of 6 °C min−1 using a TCD detector of a Shimadzu GC-14A. At the outlet of the TCD, a six-port valve sampling allowed us to analyze the gas evolved by GC using a FID detector. The sample was prepared from a CH2Cl2 solution of Sn(OtBu)4 which was contacted with pretreated silica (Aerosil Degussa 200, SSA = 196 m2 g−1) at room temperature for 2 h under stirring (Sn/SiO2 = 10 Sn wt%). The obtained slurry was filtered, the solid washed with aliquots of CH2Cl2, then dried under vacuum at 40 °C for 3 h giving a final tin loading of 4.9 ± 0.3 Sn wt%.
4.4 Catalytic experiments
Caution: since high pressures are involved, appropriate safety precautions must be taken.
The catalysts (0.100–0.550 g) were pretreated under vacuum at 130 °C for 2 h, then 20 ml of dried methanol was added. The suspension was transferred under argon into a 120 ml batch stainless steel autoclave, followed by admission of 40 g of CO2 with an ISCO 260D pump to getting a working pressure of 200 bar. The autoclave was heated to 150 °C (controlled by an internal K-thermocouple). DMC yield was analyzed by GC (Fisons 8000, FID detector) as a function of time by sampling with a high-pressure valve. At the end of the experiments, degassing the autoclave at 0 °C followed by GC analysis of the liquid phase allowed to check consistency between the two procedures; the fit was better than 3% (relative error). For recycling experiments, the catalysts were recovered by centrifugation, pretreated at 130 °C for 2 h under vacuum, then reloaded into the autoclave following the aforementioned procedure.
Acknowledgements
Financial support from CNRS-INC and the CNRS interdisciplinary program CPDD/RdR3 is greatly acknowledged. S.V. thanks the Conseil régional de Bourgogne for a postdoctoral grant.