

1 Introduction
Many efforts have been devoted for the preparation of dehydrogenation catalysts with nanoparticle structures [1]. The study of sulfated metal oxides has become an active area of research because of the high catalytic activity of organic reactions manifested by such compositions [2]. Silica/zirconia is known to have extensive catalytic activities in processes such as isomerization [1], alkylation [3], esterification [2] and those involving oxygenation of hydrocarbons [4]. The synthetic methods to such solid acids mainly differ in the type of precursor, hydrolyzing agent and sulfating agent [1–5]. Various zirconium compounds, such as Zr(NO3)4, ZrCl4, zirconium isopropoxide, zirconyl chloride, zirconium oxychloride and sometimes zirconia itself, have been used for the preparation of these solid acids [6–8]. The hydrolyzing agents such as alkaline or acidic reagents also have a significant effect on the catalyst activity. The most commonly used sulfating species are H2SO4 and (NH4)2SO4. Some sulfur compounds like H2S and SO2 have also been used [7,9]. Heterogenizing homogeneous catalysis by covalently anchored metal complexes based on chemically modified support materials have been developed these days [10,11].
Dehydrogenation reactions have been the subject of extensive studies for obtaining derivatives which can be used in pharmaceutical industries. Among the reactions which have not yet been industrially developed, dehydrogenation of 1,4-DHPs could be an interesting choice due to their antioxidant effects that may contribute to their pharmacological activities [12]. The oxidative dehydrogenation (aromatization) of 1,4-DHPs to the corresponding pyridines has been extensively studied in view of the pertinence of the reaction to the metabolism of the calcium channel blocking drugs used in the treatment of various cardiovascular disorders [13]. Furthermore, the oxidation of Hantzsch 1,4-DHPs is one of the ubiquitous issues in inorganic chemistry. In recent years several groups have reported various methods for aromatization including oxidation with ferric nitrate on a solid support [14], ceric ammonium nitrate [15], vanadomolybdo phosphate heteropolyacids [16], 4-phenyl-1,2,4-triazole-3,5-dione [17] and palladium on carbon [18]. However, most of these reactions required an extended period of time for completion, utilize strong oxidants in large excess and affording only modest yields of the products. Therefore, it is still difficult to design a catalytic oxidative dehydrogenation (ODH) system with high yield and good selectivity due to the tendency of the formed pyridine derivatives toward further oxidation. Many catalytic systems had been investigated by scientists in an attempt to find an optimum catalyst [19–21]. The most studied systems for ODH of 1,4-DHPs were based on vanadium [19] and Co complex as catalyst [22,23].
Among various transition metals, molybdenum has not previously been used in ODH reactions. Attempts to synthesize and characterize of Si-Zr-Mo nanocomposite by sol–gel method and investigation of catalytic activity toward the dehydrogenation of 1,4-DHPs both in solvent and solventless will be discussed in this presentation.
2 Experimental
2.1 Materials
All chemicals were purchased from Merck and used without further purification. Solvents were dried and distilled under nitrogen prior to use according to a standard procedure. Zirconium octanoxide was synthesized from zirconium-tetra-n-butoxide and 1-octanol by the alcohol interchange method, according to the general procedure for the preparation of metal alkoxides and was purified by vacuum distillation [24]. The reaction and manipulation of zirconium octanoxide synthesis were carried out under an atmosphere of dry nitrogen, using standard Schlenk techniques.
2.1.1 Preparation of sulfated silica-zirconia nanocomposite
The sulfated silica-zirconia nanocomposite was prepared in three steps. Initially, Zr(Oct)2SO4 was prepared by drop-wise addition of sulfuric acid (4.9 mmol, 0.26 ml, 96%) to Zr(Oct)4 solution in 0.6:1 molar ratio at room temperature. Zr-Si mixed oxide was prepared with TEOS, EtOH, AcOH and distilled water. The corresponding molar ratios of xZr(Oct)2SO4:yTEOS: 7(x + y) EtOH: 10(x + y)H2O: 2(x + y)AcOH in which the desired experiment is x = 8.2, y = 82 mmol. EtOH, TEOS, H2O and AcOH were mixed by stirring. In the next step the solution of Zr(Oct)2SO4 (0.1 M, in 82 ml 1-octanol) was added to TEOS solution under vigorous stirring [25]. The resulting sol was stirred for 3 h. The produced gel was decanted by centrifugation with 3900 r/min for 15 min and drying at 180 °C.
2.1.2 Preparation of silica-zirconia-molybdate
MoO3 (1.5 g, 7.7 mmol) dissolved in NaOH (10 mL, 5 M) was initially titrated with HCl (0.5 M) until pH = 7, followed by refluxing of the solution with sulfated silica-zirconia gel (0.5 g) for 3 h. The solid product was then decanted by centrifugation, washed with deionized water and dried at 100 °C.
2.2 Catalytic dehydrogenation
2.2.1 Preparation of 1,4-DHPs
1,4-DHPs were successfully prepared using the previously reported method [26].
2.2.2 Oxidative dehydrogenation of 1,4-DHP with H2O2
In a typical procedure, 1,4-DHP (30 mg, 0.09 mmol) and catalyst (30 mg) were added to EtOH (15 ml), H2O2 (2 ml, 30%) and the mixture was refluxed at 60 °C until the complete disappearance of 1,4-DHS based on TLC. The products were then subjected to GC and GC-MS analyses.
2.2.3 Solventless dehydrogenation of 1,4-DHPs
Dehydrogenation reactions were performed in a stirring round bottom flask fitted with a water-cooled condenser at atmospheric pressure under reflux conditions. A mixture of benzaldehyde (0.2 g, 2 mmol), ethyl acetoacetate (0.5 g, 4 mmol) and ammonium acetate (0.2 g, 3 mmol) and catalyst (0.1 g) was stirred at 90 °C until the reaction was completed. The products were extracted by ethyl acetate and dried over anhydrous Na2SO4. Two isomers of products were isolated by column chromatography on silica gel (60–120 mesh) and elution with ethyl acetate–chloroform mixture (1:4). The products were identified by GC and GC-MS analyses.
2.3 Instrumentation
Infrared spectra were performed (KBr pellets) on a Bruker Tensor 27FT-IR spectrometer. Chemical analysis of samples was carried out with Varian 150AX inductively coupled plasma (ICP) emission spectrometer. Electron microscopy was performed on a Philips EM208S, transmission electron microscope (TEM). Surface areas, pore volume and pore size distributions were obtained from the N2 isotherms which determined at 77 K using Quantachrome Nova 2200, Version 7.11 Analyzer. The products were analyzed by GC and GC-MS using Agilent 6890 Series, with FID detector, HP-5, 5% phenylmethylsiloxane capillary and Agilent 5973 Network, mass selective detector, HP-5 MS 6989 Network GC system, respectively.
3 Results and discussion
3.1 Characterization of Si-Zr-Mo nanocomposite
Si-Zr-Mo nanocomposite was prepared according to the procedure presented in Fig. 1. The exothermic reaction between sulfuric acid and Zr(Oct)4 affords the sulfated zirconia (Scheme 1, path a). Subsequent treatment with a mixture of TEOS, EtOH, acetic acid and distilled water gives the corresponding Zr-Si mixed oxide (Scheme 1, path b). Upon addition of Na2MoO4 to Zr-Si mixed oxide, Si-Zr-Mo nanocomposite is generated presumably via reaction between Zr-SO4 and MoO42− (Scheme 1, path c). In fact, the adsorption capacity of Mo to the nanocomposite depends on the number of SO42− molecules bonded to the Zr atom in the nanocomposite. The weight percentages of Si, Zr and Mo in the nanocomposite were found to be 5.69, 1.17 and 18.58, respectively. Although the amount of S in the nanocomposite before Mo adsorption was 0.4%, but no significant amount of sulphur was detected by ICP technique after Mo adsorption.
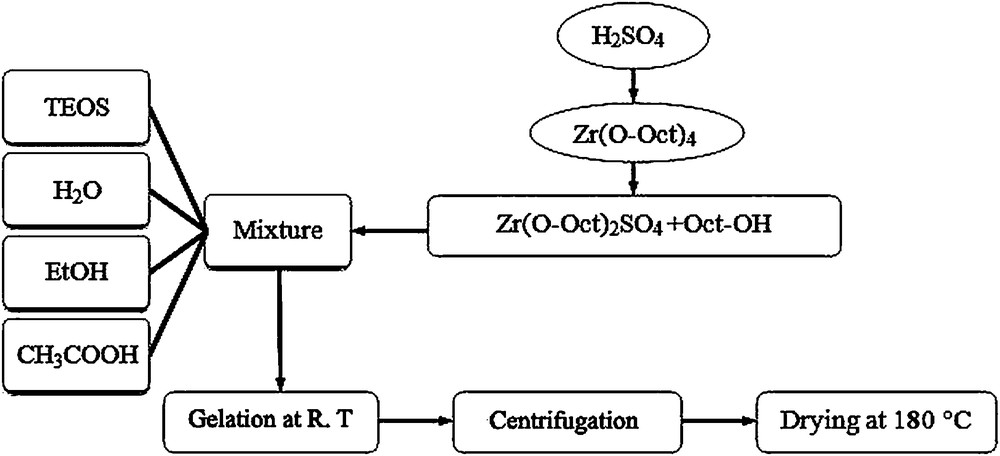
The procedure for preparing sulfated silica-zirconia nanocomposite.
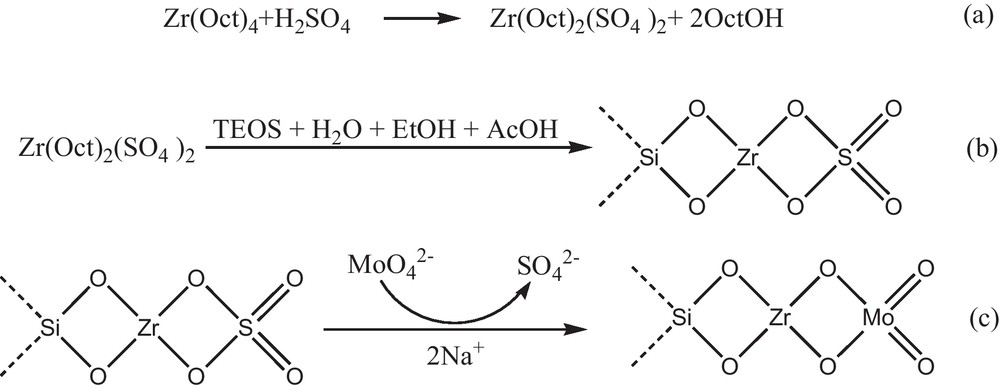
Synthesis of Si-Zr-Mo nanocomposite.
The TEM images of SO4−2/ZrO2–SiO2 and Si-Zr-Mo nanocomposite are shown in Fig. 2a, b.

The TEM image of (a) SO4−2/ZrO2–SiO2 nanocomposite, (b) Si-Zr-Mo nanocomposite.
As seen in this figure, the morphology before and after addition of MoO42− are completely different, in which for the first one is as nanoparticles but for last one as nanorods. Interestingly, different shapes of particles which are evident in the micrographs of the samples may be due to the reaction between SO42− and MoO42−, through covalent bonding, with molybdenum oxygen's and zirconium. Moreover, the amount of adsorption of Mo control the size and shape of the particles [27].
The FT-IR spectra of Zr(Oct)4, Zr(Oct)2SO4, TEOS, SO4−2/ZrO2–SiO2 and Si-Zr-Mo are shown in Fig. 3a–e, respectively, in the range of 400–4000 cm−1. No absorption was found in the range of 1750–2700 cm−1. The FT-IR spectra of zirconium octanoxide (Fig. 3a) shows vibrations at 500, 610, 725 cm−1, corresponding to Zr–O bending and stretching vibrations. The bands appearing at 1000–1300, 1300–1400 and 2800–2900 are attributed to the C–O, CH3 or CH2 and C-H stretchings of alkoxy groups, respectively. A broad band displaying at 3400 cm−1 is due to the OH stretching associated with alcohol residue present in zirconium alkoxide. The FT-IR spectrum of sulfated zirconia (Fig. 3b) indicates the vibration bands of SO42− group in the region of 900–1200 cm−1 and asymmetric and symmetric stretching vibrations of S–O bonds present in inorganic chelating bidentate sulfate [3]. Fig. 3c shows the bands displaying at 800, 1070 and 1225 cm−1 due to the stretching vibrations of Si–O and a band at 520 cm−1 belonging to bending vibration for Si–O of TEOS [2]. The broad bond appearing at 1000–1500 cm−1 indicated in Fig. 3d is probably due to vibration of Zr–O–Si present between ZrO2 and SiO2 parts [2]. As seen in Fig. 3e, the appearance of two bands at 907 and 960 cm−1 is attributed to stretching modes of Mo–O bonds of Mo particles present on the nanocomposite [28,29]. Comparison of the FT-IR spectra of Fig. 3b, d with that of Fig. 3e of the nanocomposite indicates that the MoO42− has been exchanged with SO42− anions because no vibration due to the presence of SO42− was observed. These observation are in good agreement with the ICP results.
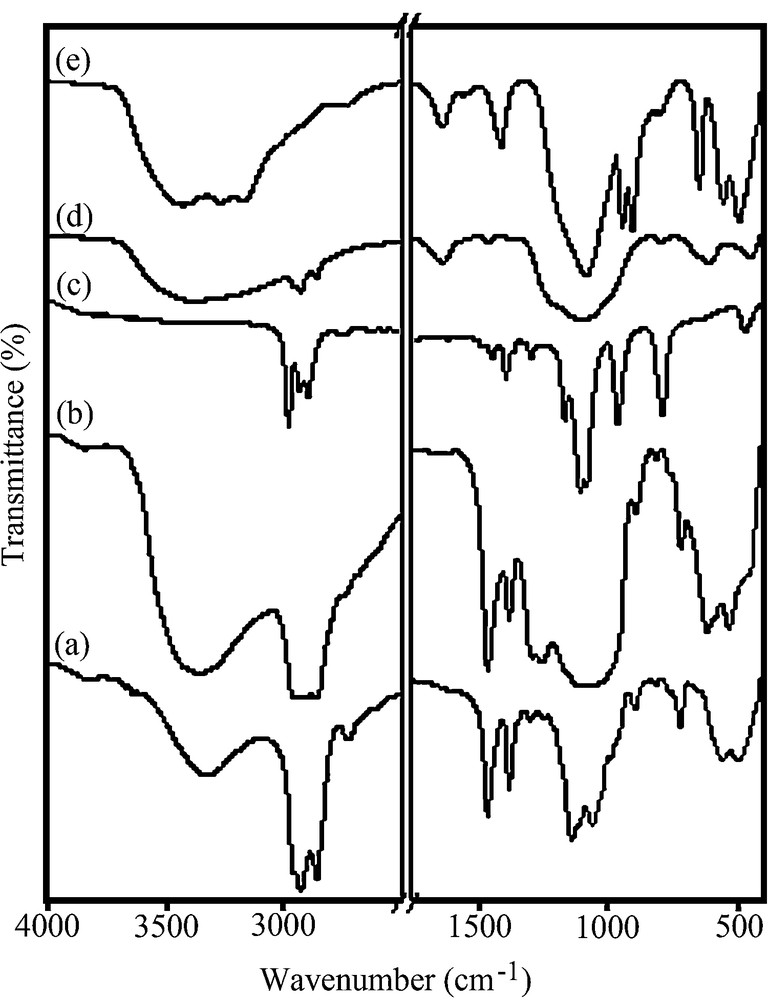
The FT-IR spectra of (a) Zr(Oct)4, (b) Zr(Oct)2SO4, (c) TEOS, (d) SO4−2/ZrO2–SiO2 nanocomposite, (e) Si-Zr-Mo nanocomposite.
The nitrogen adsorption/desorption isotherms and pore size distribution of nanocomposite are shown in Fig. 4a, b respectively. Nitrogen adsorption/desorption analyses were performed in order to investigate the textural properties of the resulting nanocomposite. The isotherm indicated a typical microporous character. The calculation of surface area and pore size distribution was based on BET and BJH method [30]. The surface area, pore size distribution, pore diameter and pore volume are 140 m2/g, 1.48 nm, 1.188 nm and 0.075 cc/g, respectively.
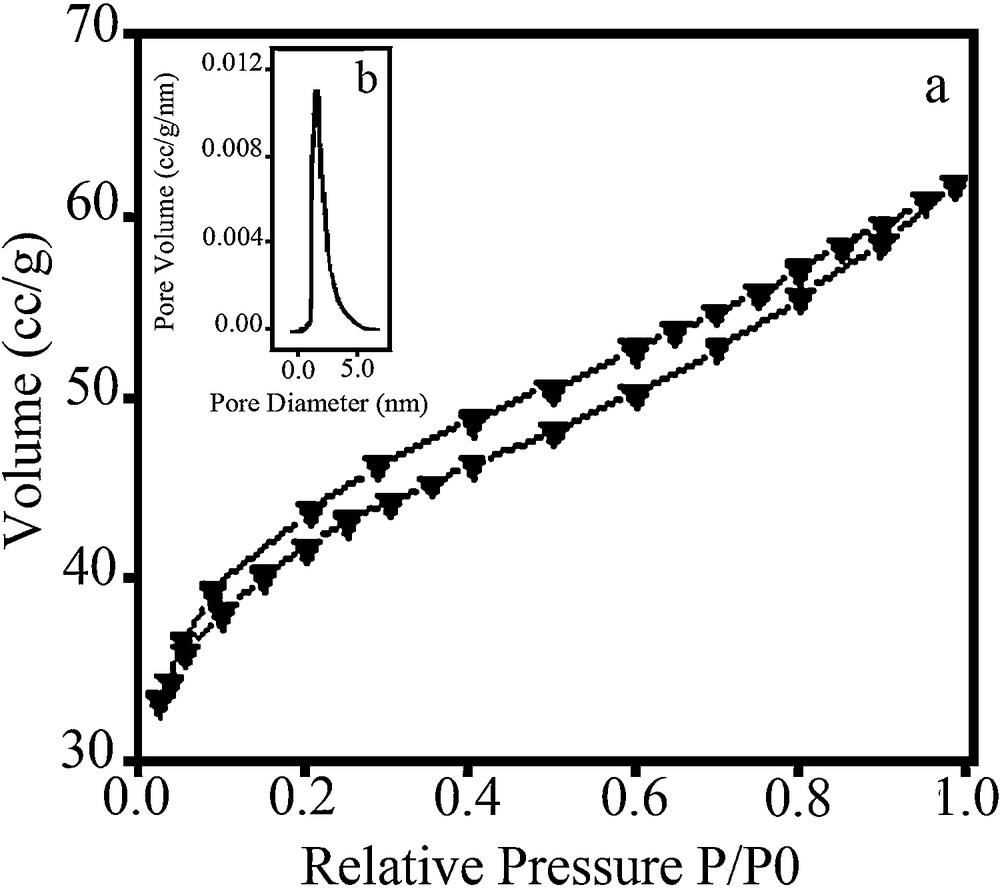
a: N2 adsorption-desorption isotherm for SO4−2/ZrO2–SiO2 nanocomposite dried at 180 °C; b: pore size distribution of SO4−2/ZrO2–SiO2 nanocomposite dried at 180 °C.
3.2 Catalytic activity
In the next step, the oxidative aromatization of the prepared nanocomposite was studied using H2O2 as oxidant and EtOH as solvent. The results of the optimization of reaction conditions based on the time and the effect of amounts of catalyst are given in Table 1. It was found that increasing the amount of catalyst from 3 mg to 30 mg increases the conversion from 40 to 100% with the formation of the corresponding pyridine derivative with 100% selectivity (entries 1–4, Table 1). The completion of reaction within 3 min seems remarkable. It was also found that increasing time of reaction from 1 to 3 minutes, the conversion increases from 45 to 100% with 100% selectivity respectively (entry 6, 5 and 4, Table 1 and Fig. 5). As seen in Table 1, 25% of DHP is converted within 600 min in the oxidative aromatization reaction in the absence of catalyst (entry 7, Table 1). Therefore, catalyst has a key role in the oxidative dehydrogenation of DHP.
The effect of reaction time and amounts of catalyst on dehydrogenation of DHP.
| Entry | Time (min) | Catalyst (g) | Conversion (%) | Selectivity (%) |
| 1 | 3 | 0.003 | 40 | 100 |
| 2 | 3 | 0.007 | 62 | 100 |
| 3 | 3 | 0.015 | 67 | 100 |
| 4 | 3 | 0.03 | 100 | 100 |
| 5 | 2 | 0.03 | 75 | 100 |
| 6 | 1 | 0.03 | 45 | 100 |
| 7 | 600 | – | 25 | 100 |
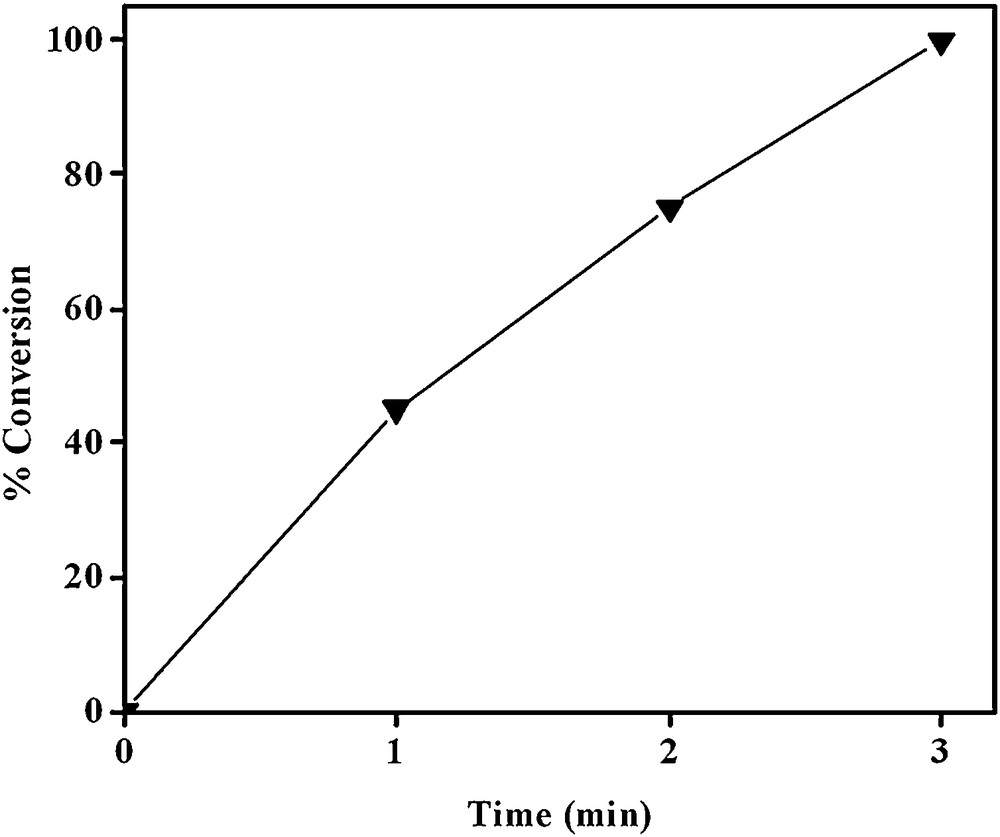
Effect of time on the aromatization of 1,4-DHP with Si-Zr-Mo nanocomposite.
The established optimized reaction conditions were then tested on DHPs with R = H, Me and Ph, prepared from suitable starting materials (Table 2). Compared to oxidative aromatization of DHP with R = Ph which proceeds to completion within 3 min (entry 3, Table 2), those DHPs with R = H and Me are quantitatively converted to the corresponding pyridine derivatives in 1 and 2 min, respectively (entries 1 and 2, Table 2). The observed rate enhancements are in accord with the decreasing steric hindrance at the substituted secondary carbon.
The effect of R group on dehydrogenation of DHP derivativesa.
| Entry | R | Solvent | Time (min) | Conversion (%) | Selectivity (%) |
| 1 | H | EtOH | 1 | 100 | 100 |
| 2 | Me | EtOH | 2 | 100 | 100 |
| 3 | ph | EtOH | 3 | 100 | 100 |
a Conditions: DHPs (0.09 mmol), catalyst (30 mg), H2O2 (1.5 ml).
The effect of other solvents on oxidative aromatization reaction of 1,4-DHPs are presented in Table 3. Compared to carbon tetrachloride as solvent, reaction has proceeded more efficiently in acetic acid (entries 2 and 3, Table 3) perhaps due to the higher solubility of the oxidant or refluxing at higher temperature. Moreover, the acid catalytic effect of this solvent on DHP containing the basic amine functional group may not be overlooked. Notably, the unsubstituted DHP proceeds much faster than DHP substituted with Ph (entries 1, 2 and 3, Table 3) [31,32].
The effect of solvent on dehydrogenation of DHP derivatives.
| Entry | R | Time (h) | Solvent | Conversion (%) | Selectivity (%) |
| 1 | H | 12 | CCl4 | 97 | 100 |
| 2 | Ph | 24 | CCl4 | 19 | 100 |
| 3 | Ph | 24 | CH3COOH | 50 | 100 |
The solventless dehydrogenation of 1,4-DHPs was carried out using Si-Zr-Mo nanocomposite based on the Scheme 2, with benzaldehyde, ethyl acetoacetate and ammonium acetate as starting materials at 90 °C.
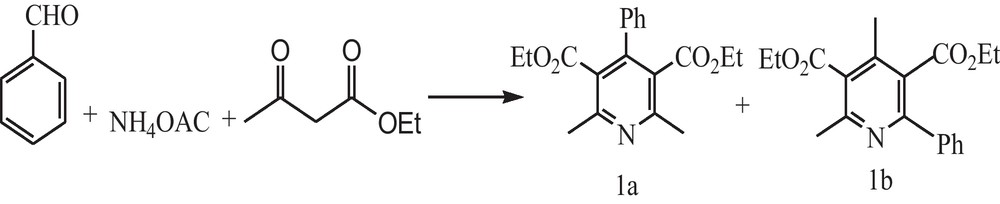
Synthesis of conventional dihydropyridines in the presence of Si-Zr-Mo nanocomposite under reflux condition.
In order to optimize the reaction conditions, the effect of the amount of catalyst and reaction time was studied (Table 4). As illustrated in this table, increasing the amount of catalyst from 30 mg to 100 mg the conversion has been increased from 53 to 92% with selectivity 80% and 20% toward the formation of 1a and 1b respectively (Table 4, entries 1–3). The reaction time was also extended to 15, 18, 21, and 24 h (Table 4, entries 3–6) with conversion 48, 65, 78 and 92% respectively.
Condensation of benzaldehyde, ethyl acetoacetate and ammonium acetate catalyzed by nanocomposites under different times of reaction and different amounts of catalyst in solventless condition.
| Entry | Catalyst (mg) | Time (h) | Conversion (%) | Selectivity (1a %) | Selectivity (1b %) |
| 1 | 30 | 24 | 53 | 47 | 47 |
| 2 | 70 | 24 | 80 | 63 | 37 |
| 3 | 100 | 24 | 92 | 80 | 20 |
| 4 | 100 | 21 | 78 | 77 | 23 |
| 5 | 100 | 18 | 65 | 77 | 23 |
| 6 | 100 | 15 | 48 | 67 | 33 |
A possible mechanism of the Si-Zr-Mo catalyzed aromatization of 1,4-DHPs with H2O2 is suggested in Scheme 3. Initially, coordination of H2O2 with Mo of Si-Zr-Mo nanocomposite I affords the corresponding proxo intermediate II [29]. As indicated in Scheme 3, a concomitant proton and hydride transfer from N-H and C-H of 1,4-DHP to nucleophilic M = O and electrophilic HO-O oxygen's of II, respectively leads to the corresponding substituted pyridine and regeneration of the catalyst via elimination of water [19].
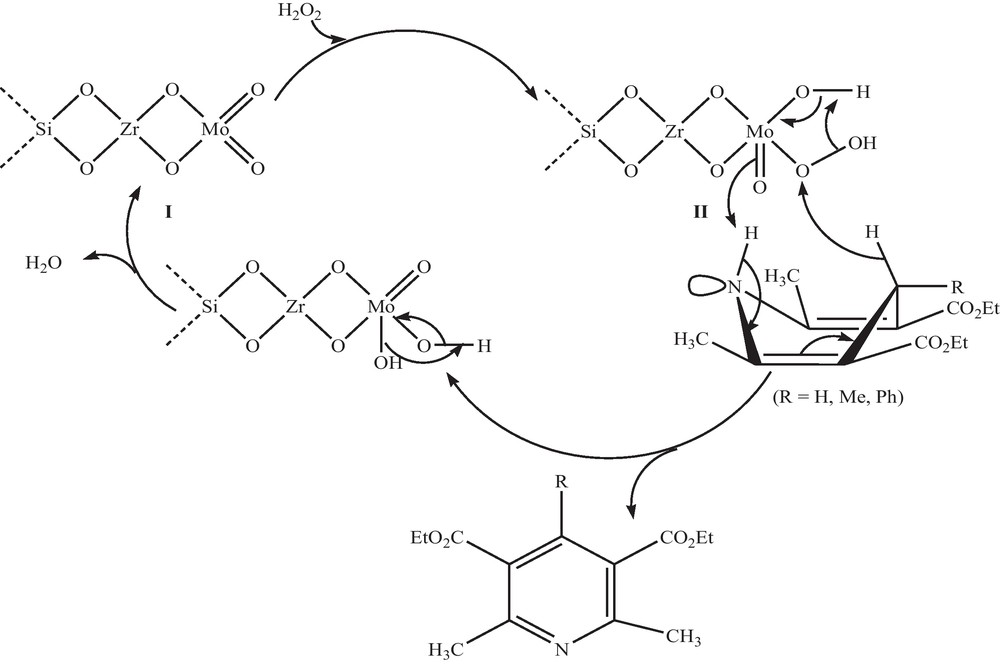
Proposed mechanism for the oxidative aromatization of Hantzsch 1,4-DHP with Si-Zr-Mo nanocomposite.
4 Conclusion
The Si-Zr-Mo nanocomposite was prepared using Zr(Oct)4, H2SO4, TEOS and sodium molybdate via sol–gel method. The nanocomposite with surface area, pore size, pore diameter and pore volume of 140 m2/g, 1.48 nm, 1.188 nm and 0.075 cc/g, respectively was obtained. This work showed that the addition of Mo plays a crucial role on the particle size and morphology of the nanocomposite. The prepared nanocomposite was tested as catalyst for oxidative aromatization of 1,4-DHPs. A number of 1,4-DHPs were quantitatively converted to the aromatic analogous within 1 to 3 min. The attractive features of this protocol are a simple reaction procedure, short reaction time, easy product separation and purification.
Acknowledgment
The authors would appreciate the Alzahra University and Agricultural, Medical and Industrial Research School (AMIRS) for supporting this work.