

1 Introduction
Polyoxometalate (POM) chemistry represents a large established area in inorganic chemistry [1,2], and the ongoing interest is driven mainly by its potential applications in (electro) catalysis [3], biology [4] and more recently nanochemistry [5]. The properties of such a class of molecular materials are intimately related to their structural architecture and composition. Thus, the possibility to design new architectures and thereby to increase the library of available topological structures is an important goal in today's chemical research. Actually, this wide class of compounds is mainly dominated by a few structural archetypes, namely the Linqvist, Keggin and Dawson anions [6]. These archetypes have been derived as polyvacant species at the origin of a large series of mixed metal compounds. Thus, vacant polyoxotungstate ions constitute a remarkable class of rigid and robust ligands convenient for predictable coordination chemistry [7,8]. Such a class of anionic ligands ranging from monovacant to hexavacant exhibits various polyhedral arrangements mainly derived from the most common archetypical structural motifs i.e. Dawson or Keggin structures [9]. The number of possible combinations is further increased by the possibility of various types of isomerism e.g. the Baker-Figgis isomers in the Keggin series (α, β, γ, δ and ɛ) [10]. In this context, the chemistry of polyvacant heteropolytungstates and the understanding of the related self-assembly are still a challenge and represent potential openings for the discovery of more and more finely designed forthcoming mixed metal compounds. Schematically, Keggin derivative heteropolyoxotungstates can be distributed into two subclasses, one built from tetrahedral assembling groups {XO4} with X = PV, AsV, SiIV, BIII, the other derived from {XO3} trigonal assembling groups with X = AsIII, SbIII, TeIV. The stereochemistry of this last class of heteropolyoxotungstates is mainly dominated by the presence of the quite inert electronic lone-pair located at the heteroatom X, and the numerous species obtained from these lacunary subunits retain the B-type isomerism. [11] As usual in POM chemistry, the formation of these species is governed by a complex interplay involving the various main parameters, such as pH, W/As ratio, nature of the starting materials, counter ions, temperature and ionic strength. A series of sandwich-type anions has been isolated, which consists of two [B-AsW9O33]9− subunits linked by three, two or one oxotungstic {WO}4+ cationic linkers. As expected, the formation of these three related compounds is mainly pH dependant. The metal saturated anion [As2W21O69(OH2)]6− is obtained in strongly acidic medium [12] and can be prepared even as an acid through diethylether-hydrochloric acid extraction (Drechsel method) [13]. The monovacant derivative [As2W20(OH2)2O68]10− is obtained at pH∼2 while the divacant [As2W19(OH2)O67]14− anion is formed at pH ∼ 6 [14,15]. It has to be noted that the formations of the mono- and divacant anions require the presence of potassium ions within the synthetic medium. For such sandwich-type arrangements, the stabilizing role of the potassium cations is now well established and can be understood merely from its ionic radius which perfectly matches with the size of the anionic vacancies. In the presence of the sodium cation at pH = 4, the WO42−/As2O3 chemical system led to the crown-shape anion [As4W40O140]28− previously reported by Hervé et al. [16] while the use of the preformed K14[As2W19(OH2)O67] compound at pH ∼ 2 led to the largest tungsto-arsenate [As6W65(OH2)7O217]26− anion [17]. This compound presents the peculiarity of containing both the α and β isomers derived from the [B-AsW9O33]9− trivacant subunit bridged together through cis-{WO2}2+ and trans-{WO(OH2)}4+ linking groups [17]. This subclass of polyoxotungstates has been widely extended by including transition metal cations in their molecular framework. Many of them derive from the lacunary tungstoarsenate precursors mentioned above [18] but the most spectacular mixed metal representative is certainly the macrocyclic anion [As12Ce16(OH2)36W148O524]76− which corresponds to the largest polyoxotungstate known to date [19]. We report herein on the synthesis of a new {Mo2O2S2}-supported tungstoarsenate arrangement which involves tungstate [WO4]2−, preformed trivacant anion [α-B-AsW9O33]9− and the oxothio dication [Mo2O2S2(OH2)6]2+. The [As3W30(OH2)3O102]15− backbone is formed only in the presence of the dioxothiocation [Mo2O2S2(OH2)6]2+ and potassium ions, both located within the central anionic cavity of the {As3W30(OH2)3O102}15− macrocyclic moiety. The resulting anion (1) has been structurally characterized by single crystal X-ray diffraction study and 183W NMR in solution.
2 Result and discussion
2.1 Structural description of the anion
The molecular structure of 1 is depicted in Fig. 1 and the crystallographic data are summarized in Table 1. The anionic molecular arrangement of the anion 1 is based on the tungsto-arsenate(III) backbone, which consists of three [α-B-AsW9O33]9− subunits linked together by three {O = W–OH2}4+ groups forming a triangular arrangement (Fig. 1a). The oxo and aquo ligands of the three {O = W–OH2}4+ linkers are arranged in a trans disposition (Fig. 1a) where the W = O double bonds of the three {O = W–OH2}4+ linkers are clearly identified by their short W–O separations (1.674–1.712 Å) while the trans W–OH2 groups exhibit longer W–O distances (2.393–2.484 Å). Furthermore, the three {O = W–OH2} are disposed equally with their W = O bonds pointing toward the centre of the cluster. The geometrical parameters (bond distances and angles) observed within the tungsto-arsenate molecular framework fall in the range that is usually reported for such kind of structures. The tungsto-arsenate(III) backbone has retained the [As3W30(OH2)3O102]15− composition (abbreviated {As3W30} hereafter). Such an arrangement delimits a large inner cavity lined by the remaining six terminal oxygen atoms belonging to the three [AsW9O33]9− subunits. Interestingly, the structure of 1 reveals the presence of the {Mo2O2S2(OH2)4}2+ core, grafted on the polyoxoanion surface and spanning two adjacent [AsW9O33]9− subunits (shown in Fig. 1b). Each Mo(V) atom appears similarly bound through a single Mo–O–W bridge, wherein the bound oxygen atom of the Mo coordination sphere occupies an equatorial position. Besides, the coordination sphere of the Mo(V) atoms is filled by two aquo ligands to give the typical {MoS2O4} octahedral arrangements. The Mo–O bond distances fall in the 2.182–2.203 Å range for equatorial aquo ligands and in the 2.431–2.460 Å range for axial aquo ligands. Interestingly, these attached water molecules interact through an internal hydrogen bond network with terminal oxygen atoms belonging to the {As3W30} framework (Fig. 1b). Each equatorial water molecule interacts with the closest neighboring terminal oxygen atom to give a short hydrogen bonding contact (O•••O = 2.646(1)–2.683(2) Å), whereas the two axial water molecules appears hydrogen bonded with the same terminal oxygen atom, belonging to the {O = W–OH2}4+ linker (O•••O = 2.882(2)–2.918(2) Å). Then, the triangular tungsto-arsenate(III) backbone delimits a large anionic open cavity, partially filled on one side by the {Mo2O2S2(OH2)4}2+ cation. The remaining void of the cavity is occupied by the potassium cation labeled K1, which appears closely embedded between two inner aquo ligands of the {Mo2O2S2(OH2)4}2+ core (K–O = 2.916–2.943 Å) and four terminal oxo groups, two belonging to one {AsW9O33} subunit (K–O = 2.792–2.816 Å) while the other two pertain to the vicinal {O = W–OH2}2+ linkers (Fig. 1c). Furthermore, two additional oxo ligands supplied by a nearby {As3W30} unit complete the coordination sphere of the potassium ion to give it a eight-fold coordination. Two other potassium ions, labelled K2 and K3, are symmetrically linked to {AsW9O33} subunits and located around the central cavity delimited by the {As3W30} backbone (Fig. 1c). These potassium ions are bound to six oxygen atoms belonging to the adjacent {AsW9O33} subunits, and distributed as two terminal and four bridging oxygen atoms with K–O distances which fall in the 2.869(9)–3.210(10) Å range. One tetramethyl ammonium cation has been revealed in the structure of 11, in the close vicinity of the {As3W30} backbone on the opposite face where the {Mo2O2S2} core and the three embedded potassium ions are located (Fig. 2). Such a cation is found on this side, the triangular {As3W30} arrangement designs the contour of a small anionic pocket, filled by one methyl group of the NMe4+ ion. The analysis of the cell packing reveals that close interactions exist between two anions 1, symmetrically arranged to form a large dimeric association (Fig. 3). Within the dimer, both anions are related through an inversion center and interact together through intermolecular contacts such as ionic interactions through four O–K–O bridges involving the three potassium ions labelled K1, K2, K3 and through four short hydrogen bonds spanning the two equatorial aquo ligand attached to each {Mo2O2S2} groups and bridging oxygen atoms of the adjacent {As3W30} unit. Thus, the cohesion of the dimer is ensured by four O–K–O junctions involving K–O distances in the 2.68–2.90 Å range and four hydrogen bonds with O•••O distances between 2.64(1) and 2.70(1) Å. As observed experimentally, the anion 1 displays no local symmetry in the solid state, while idealized Cs symmetry can be approximated for the isolated species 1, and C2h symmetry for the geometrically idealized dimeric arrangement (both monomers are related by an inversion center).
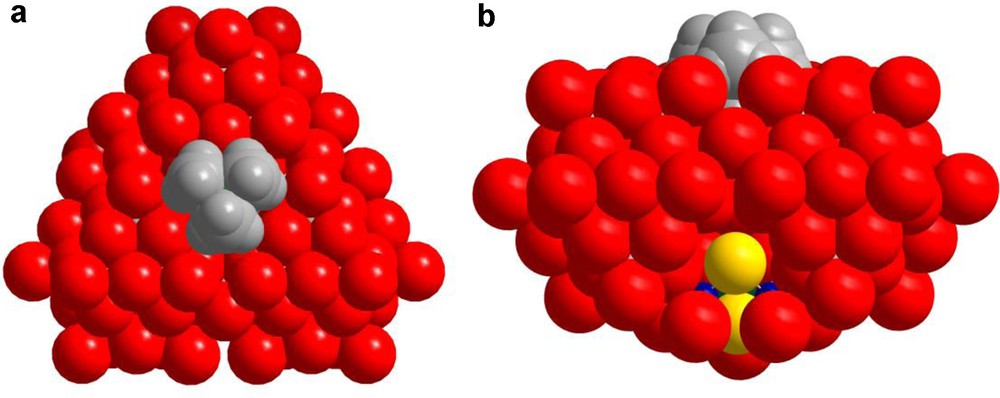
Space filling representations highliting the close interactions between the NMe4+ cation and the anion 1: (a) top view showing the location of the NMe4+ cation at the centre of the triangular arrangement. (b) side view showing the partial inclusion of the NMe4+ within the anionic pocket.
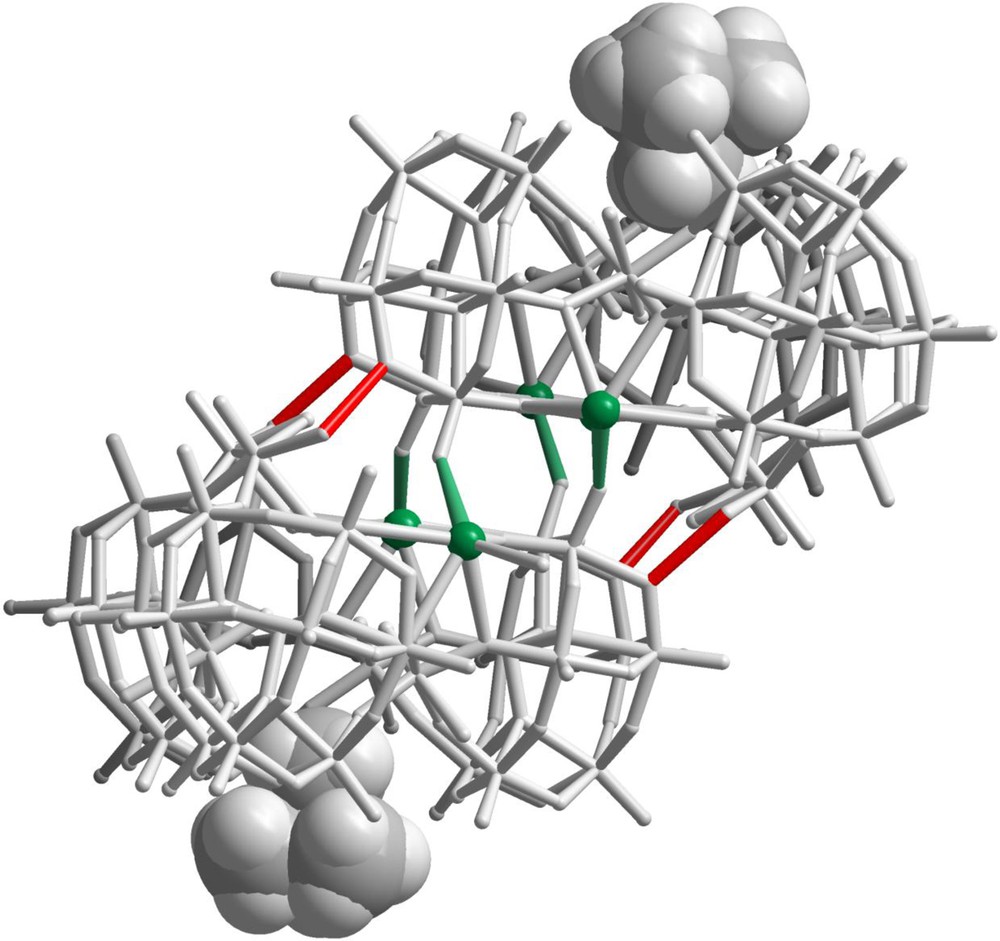
Representation showing of the association of two anion 1 through four hydrogen bonds (red stick) and four K–O bonds (green stick) involving the K2 and K3 cations. The two interacting NMe4+ cations are represented by space-filling model (grey spheres).
2.2 183W NMR in solution
The 183W NMR spectrum of the anion 1 as Li+ salt is shown in Fig. 4. It consist of 16 lines with intensity ratios of 2:2:2:2:2:2:2:2:2:1:2:2:2:1:2:2, corresponding to the 30 W nuclei involved within the structure. The Cs symmetry for the isolated anion 1 generates two equivalent {AsW9} subunits which have no local symmetry, giving rise to nine resonances of equal intensity ratios of 2. The remaining {AsW9} crossed over by the symmetry plane leads to five resonances with intensity ratios 2:2:2:2:1, while the three bridging {O = W–OH2} group give two lines of 2:1 intensity ratio. The chemical shifts of the sixteen resulting lines observed between δ = −50 and −200 ppm fall in the expected range for WVI nuclei involved within an octahedral oxo environment. However, no further interpretation, such as a partial assignment, can be given since the close proximity of many resonances prevents the determination of the W–W coupling satellites. Nonetheless, the 16 183W NMR line spectrum is fully consistent with the solid state structure of the anion 1, thus confirming the stability of the anion as Li salt in solution.
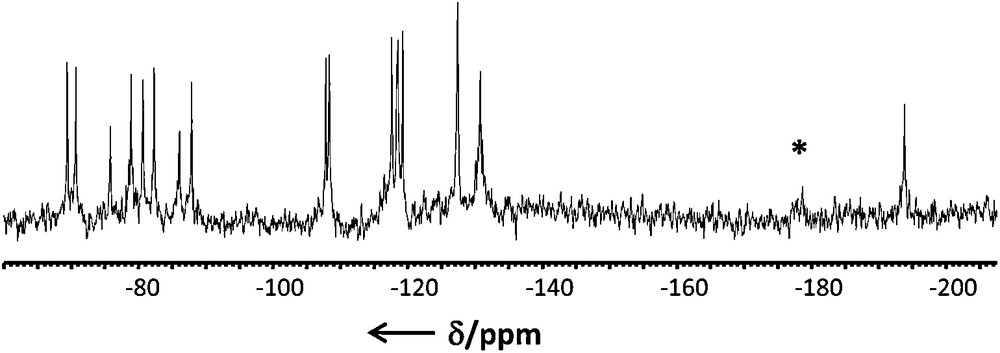
183W NMR spectrum of the anion 1 as Li salt in aqueous solution (*: unidentified impurities).
2.3 Synthesis of 1
1 was obtained from a three component synthetic procedure, using [Mo2O2S2(OH2)6]2+ obtained from the acidic hydrolysis of the cyclic precursor K2-x(NMe4)x[I2Mo10O10S10(OH)10(H2O)5]•20H2O, Na2WO4 and the mixed sodium-potassium salt K6Na3AsW9O33•6H2O with ratios close to the expected stoichiometry. The mixing sequence of the three reactants appears crucial for the formation of 1. The first step corresponds to the addition of [WO4]2− to a 2 mol L−1 KCl and 0.1 mol L−1 HCl solution containing the [Mo2O2S2(OH2)6]2+ oxothiocation. The cloudy yellow solution probably results from the formation of polymeric compounds due to condensation processes between the acidic cation {Mo2O2S2}2+ and the basic anion [WO4]2−. The solution turns red after addition of the [AsW9O33]9− anion and becomes clear at pH = 1.4 under moderate heating. Changing the sequences order of reactant addition does not lead to any crystalline materials, but results systematically in an abundant amorphous precipitate. Furthermore, the structural analysis of 1 allows one to hypothesize that the oxotungstic framework {As3W30} anion could exist as isolated species, thus completing the {AsxW10x} series (with x = 2 or 4). Similar syntheses carried out without {Mo2O2S2}2+ in similar conditions of pH, reagent concentrations, ionic strength and temperature give colorless well-shaped crystals. Nevertheless, the 183W NMR characterization of this crystalline material as Li salt gives a complex spectrum containing more than 40 resonances between −80 and −210 ppm, exhibiting a wide distribution of intensity ratios (not shown). Such a result is obviously inconsistent with the [(α–AsW9O33)3(WO(OH2))3]15− anion, characterized in 1. In conclusion, the formation of 1 needs the simultaneous presence of WO42− and {Mo2O2S2}2+ during the self-assembly processes. Furthermore, although the {Mo2O2S2}2+ core (and also potassium ions) appear to be weakly bound to the [(α–AsW9O33)3(WO(OH2))3]15− backbone within the resulting anion 1, this cation i.e. {Mo2O2S2}2+ plays a crucial role in the self-assembly processes, thus justifying the used statement “{Mo2O2S2}2+ directed synthesis”.
3 Experimental
3.1 Physical methods
Water content was determined by thermal gravimetric analysis (TGA7, Perkin-Elmer).
Elemental analyses were performed by the service central d’analyse du CNRS, Vernaison, France.
Infrared spectra were recorded on a Magna 550 Nicolet spectrophotometer, using KBr pellets.
3.1.1 183W NMR measurements
The 183W spectrum was recorded at 20 °C in 10 mm o.d. tubes on a Bruker Avance-300 FT-NMR spectrometer operating at 12.5 MHz. Chemical shifts were referenced to an external 2 mol L−1 Na2WO4 solution in alkaline D2O and to an external secondary reference of SiW12–D2O solution (δ = −103.8 ppm). The saturated aqueous solution of Li13[(α-AsW9O33)3(WO(OH2))3(Mo2O2S2(H2O)4)]•20H2O was obtained by cationic exchange of the corresponding potassium salt through a Dowex 50W-X2 resin (Li+ form). The eluate was evaporated until dry and dissolved in a mixture of H2O-D2O (v/v) to obtain a concentration of about 2.6 mol L−1 in W. 183W NMR of the Li13-1 in D2O/H2O (v/v)/ppm: −194,6 (2); −131,8 (2); −128,1 (2); −120 (2); −119,3 (2); −118,3 (2); −109,1 (2); −108,5 (2); −88,6 (2); −86,7 (1); −83,1 (2); −81,3 (2); −79,7 (2); −76,4 (1); −71,4 (2) −70 (2).
3.1.2 X-ray diffraction studies
A well-shaped orange parallepipedic crystal (0.16 × 0.12 × 0.06 mm3) of (NMe4)K12As3W30Mo2O2(OH2)7O104•20H2O (noted TMAK12-1) was mounted in sealed Lindeman tubes to prevent any loss of crystallization water. Intensity data collections for TMAK12-1 were carried out at room temperature on a three-circle Siemens SMART diffractometer equipped with a CCD bi-dimensional area detector and using a normal-focus molybdenum-target X-ray tube (= 0.71073 Å). An empirical absorption correction based on the method of Blessing [20] was applied by using the SADABS program [21]. The structures were solved by direct methods and refined by the full-matrix least squares method by using the SHELX-TL package [22]. Relevant crystallographic data for compounds TMAK12-1 are reported in Table 1. Heavier atoms (tungsten and molybdenum) were initially located by direct methods. The remaining non-hydrogen atoms were located from Fourier differences and were refined with anisotropic thermal parameters. The disordered atoms, potassium cations and oxygen atoms of crystallized water were isotropically refined. Among the 12 potassium cations and 20 crystallized water molecules, only five and 10 were located, respectively, probably due to severe disorders, a common feature in POM structures. Further details on the crystal structure investigation may be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49 7247 808 666; e mail: crysdata@fiz karlsruhe de, on quoting the depository number CSD 423110 for TMAK12-1).
3.2 Syntheses
The precursors K2−x(NMe4)x[I2Mo10O10S10(OH)10(H2O)5]•20H2O (0 < x < 0.5) were prepared as described in literature [23]. The mixed salt K6Na3[AsW9O33]•12 H2O was obtained from a procedure derived from that published by Tourné et al. for the sodium salt Na9[AsW9O33]•19.5 H2O [24]. Both precursors have been characterized by routine methods (FT-IR, EDX, TGA).
K6Na3[AsW9O33]•12 H2O. Sodium tungstate (60 g; 0.18 mmol) and sodium metaarsenite (2.6 g; 20 mmol) are dissolved in 100 mL of boiling water. The pH is down to about 7 by slow addition of 17 mL of concentrated hydrochloric acid. After 1 h, potassium chloride (22 g; 295 mmol) is added to the hot solution provoking the precipitation of a white abundant solid. After cooling at room temperature, the solid is collected by filtration and dried with ethanol and diethyl ether. Yield: 95% based on W. Elemental analysis calcd (%) for H24K6Na3AsW9O45 (M = 2777.17 g.mol−1): As 2.69; K 8.44; Na 2.48; W 59.58. found: As 2.44; K 8.68; Na 2.37; W 58.12. IR/cm−1, (KBr pellet): 932(m), 895(s), 777(s), 731(s), 600(vw), 508(w), 470(w), 437(w), 412(w), 354(w), 340(sh).
NMe4K12[(α-AsW9O33)3(WO(OH2))3(Mo2O2S2(H2O)4)]•20H2O, TMAK12-1. K2-x(NMe4)x[I2Mo10O10S10(OH)10(H2O)5]•20H2O (0.812 g, 0.35 mmol) was dissolved in 0.1 M HCl solution (170 mL) containing 2 M of KCl. Na2WO4.2H2O (1.25 g, 3.79 mmol) was added to the previous red-orange solution, leading to a cloudy yellow solution. K6Na3[AsW9O33]•12H2O (10 g, 3.74 mmol) was finally added to the solution, giving a red-orange solution. The pH was adjusted to 1.4 by few drops of HCl 1 M. Solution was heated to 35–40 °C under stirring until becoming limpid. This mixture was kept to slowly evaporate at room temperature in air. After 5 days, 1.1 g of parallepipedic orange single crystals were obtained and isolated by filtration (yield 10% based on W). Elemental analysis calcd (%) for H66NC4K12As3W30Mo2S2O131 (M = 8689.46 g.mol−1): As 2.58; C 0.55; K 5.39; Mo 2.20; N 0.16; S 0.73; W 63.47. found: As 2.67; C 0.71; K 6.02; Mo 2.13; N 0.23; S 0.79; W 62.73. IR/cm−1, (KBr pellet): TMA+: 1482(w), 964(s), 903(s), 826(w), 764(w), 733(w), 482(w).
1 The tetramethyl ammonium cations are provided by the {Mo2O2S2}-containing precursor which corresponds to the mixed tetramethyl ammonium-potassium salt K2-x(NMe4)x[I2Mo10O10S10(OH)10(H2O)5]·20H2O (0 < x < 0.5).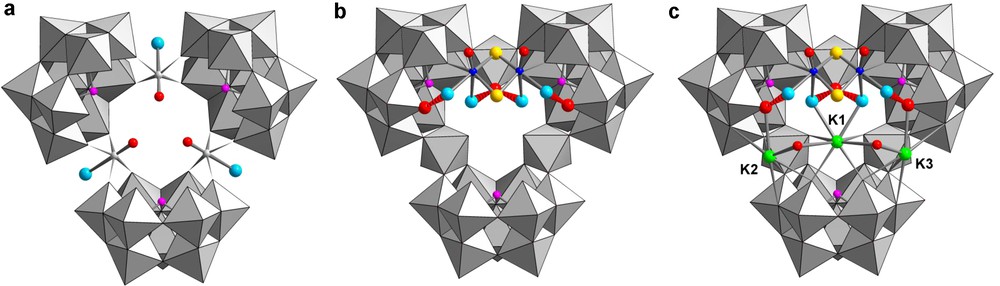
| Formula | H66NC4K12As3W30Mo2S2O131 | ρcalc, g cm−3 | 3.513 |
| M, g mol−1 | 8689.46 | μ(MoKα), Å | 22.697 |
| Crystal size (mm) | 0.16 × 0.12 × 0.06 | λ(MoKα), Å | 0.71073 |
| Space group | P–1 | θ range, deg | 0.95/25 |
| a, Å | 20.0217 (17) | Data collected | 54202 |
| b, Å | 20.7097(14) | Unique data | 27587 |
| c, Å | 21.7790 (14) | Unique data I > 2σ(I) | 20409 |
| α, deg | 84.118 (4) | Parameters number | 1475 |
| β, deg | 79.186 (3) | R1(F)a | 0.0486 |
| γ, deg | 64.225 (2) | wR2 (F2) | 0.1538 |
| V, Å3 | 7985.4 (10) | GOF | 1.165 |
| Z | 2 |