

1 Introduction
Many heterocyclic compounds bearing butenolides and lactam scaffolds are of synthetic interest, since they display extensive biological activities [1,2], and constitute an important class of natural (Fig. 1) [3–6] or unnatural products [7–10]. Moreover, they are extremely versatile building blocks for the manufacture of bioactive compounds in pharmaceutical drug design and the agrochemical industry. Nowadays, more and more natural products containing butenolides and lactam ring systems have been isolated from a variety of marine and terrestrial organisms, such as bacteria, moulds, algae, fungi, lichens, and sponges [11,12]. On the other hand, many butenolides and lactam derivatives featuring 3-acyl substituents are novel ligands for their strong ability to chelate biologically important metal cations and to mimic phosphate groups in the binding site of phosphatases and kinases [13–15]. Very recently, some butenolides and lactam derivatives are also widely applied as versatile building blocks for the construction of novel heterocycles and fused heterocycle system [16–19].

Representative structures of naturally occurring butenolides and lactam derivatives.
Thus, development of novel functionalized multi-substituted butenolides and lactam heterocyclic derivatives as pharmaceuticals is still an important area of interest in life sciences, and the search for an efficient method for the construction of these heterocyclic derivatives under mild conditions is not only highly desirable but also necessary. The promising bioactive and structural diversity of these classes of heteroaryl compounds urged us to conveniently construct a series of novel structural variants of multi-substituted butenolides and lactam derivatives and their related intermediates. In this short communication, we would like to present a molecular diversity-oriented convenient procedure for large-scale access to construct bioactive butenolides or lactam units (Fig. 2) that exploit mild ring-closure methods and cheap raw materials, which will have widely applied prospects because of its easy to obtain raw material and its convenient operation.
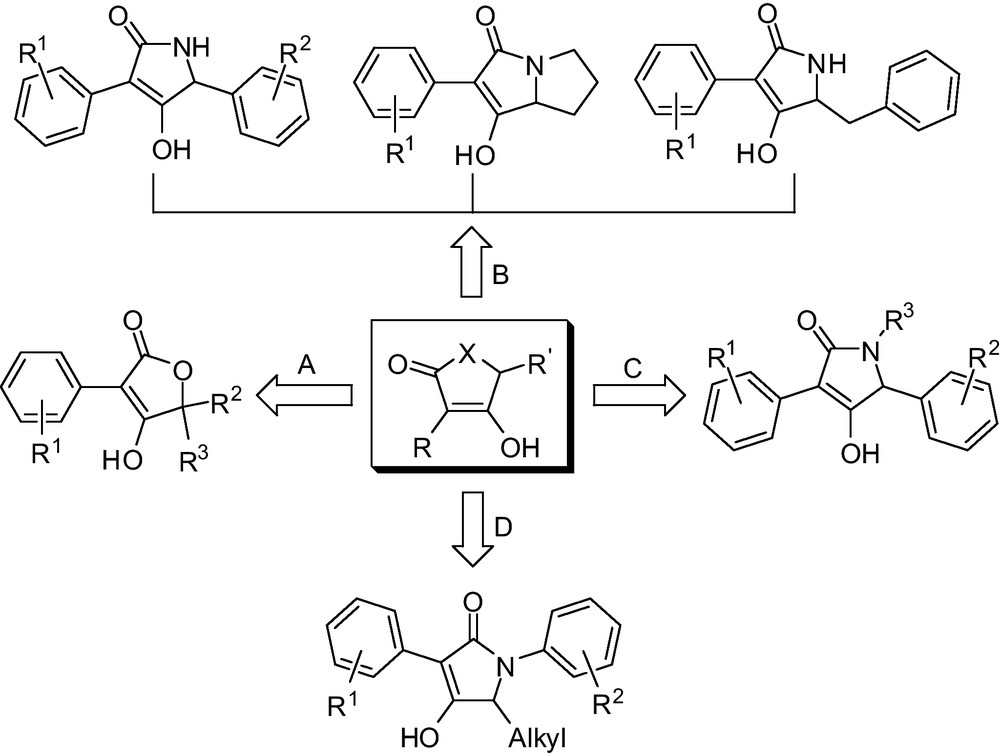
Design strategy for polysubstituted butenolides and lactams.
2 Results and discussion
2.1 Synthesis of multi-substituted butenolides derivatives
Considering the convenient synthesis to construct the structural diversity of multi-substituted butenolides and lactam scaffolds, various amino-acids, α-hydroxy acids and suitable phenylacetic acids were selected as raw materials. Polysubstituted butenolides derivatives 5 were prepared in a simplified and more efficient method, which is outlined in Scheme 1.
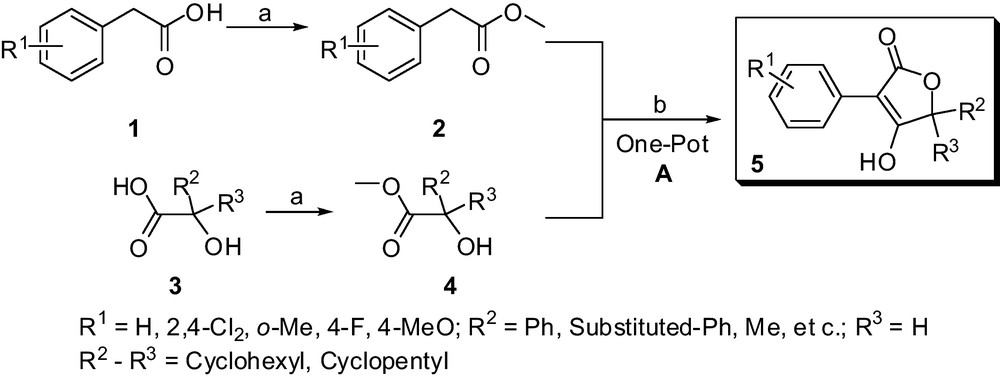
Method A. Synthesis of multi-substituted butenolides derivatives 5. Reaction conditions: a: MeOH, Conc. H2SO4, reflux 6 to 8 h; b: tBuOK or EtONa, THF, reflux for 8 to 10 h.
For the sake of structural diversity, various easily available α-hydroxy acids such as lactic acid, 1-hydroxycyclohexanecarboxylic acid, mandelic acid and their analogues can be selected as raw materials. As shown in Scheme 1, the target butenolides derivatives 5 were conveniently obtained via two steps including esterification, one-pot heterocyclization reactions. The cyclization reaction was accessed conveniently by the modified one-pot heterocyclization reaction [20,21] from various substituted methyl 2-phenylacetate 2 and methyl 1-hydroxycarboxylate 4 in good yields (Table 1), which involve a transesterification step, followed by the Dieckmann condensation. According to the above-described experimental conditions, the butenolide derivatives 5 were easily achieved with a high yield and significantly simple disposal compared to the reported chromatographic methods [20,21]. Nowadays, several butenolide derivatives could be obtained for large-scale production (> 500 g) without column chromatography in our laboratory.
Synthesis of multi-substituted butenolides derivatives 5.
| Entry | Compd. | Substituents | Appearance | Yield (%)a | ||
| R1 | R2 | R3 | ||||
| 1 | 5a | 4-MeO | -(CH2)5- | White powder | 75 | |
| 2 | 5b | 4-F | H | Ph | Light yellow powder | 75 |
| 3 | 5c | 2,4-Cl2 | H | Ph | Light yellow powder | 78 |
| 4 | 5d | 4-MeO | H | Ph | White powder | 71 |
a Yields refer to isolated products.
2.2 Synthesis of multi-substituted lactam derivatives
Scheme 2 describes the outline for the construction of representative multi-substituted lactam compounds 12 and 13, which were conveniently obtained via esterification, acylation, N-alkylation and intramolecular heterocyclization reactions. The cheap amino-acids such as phenylglycine, phenylalanine, proline and its analogues were all selected as starting materials, which were conveniently converted to the various amino-acid ester hydrochlorides 8 under mild conditions. Subsequently, the compounds 8 are easy to transfer to the N-substituted amino-acid ester 9 via N-alkylation reactions. The key intermediate phenylacetamide derivatives 10 and 11 were accessed conveniently from intermediates 8, 9 and various substituted 2-phenylacetyl chlorides, respectively. The following intramolecular heterocyclization reaction was treated in the presence of potassium tert-butoxide, and the several kinds of target compounds (Method B in Fig. 2) were achieved with a simple disposal and high yields, which may be propitious to large-scale production. All newly synthesized compounds were characterized as 4-hydroxy-3-substituted phenyl-1H-pyrrol-2(5H)-one derivatives on the basis of satisfactory analytical and spectral data including 1H NMR, 13C NMR, ESI-MS, IR and element analyses. Mass spectra, in particular, showed the molecular ion peaks at the appropriate m/z values. The 1H NMR spectroscopic data are in agreement with the proposed structures. The general 1H NMR spectra of multi-substituted butylene lactam derivatives 12 show obvious singlet peak in the range 11.20-11.40 ppm assigned to the protons of the hydroxy linked to lactam heterocycle. The signals in the range 4.10-5.07 ppm indicated the corresponding methine protons next to the N atom in the heterocycle.
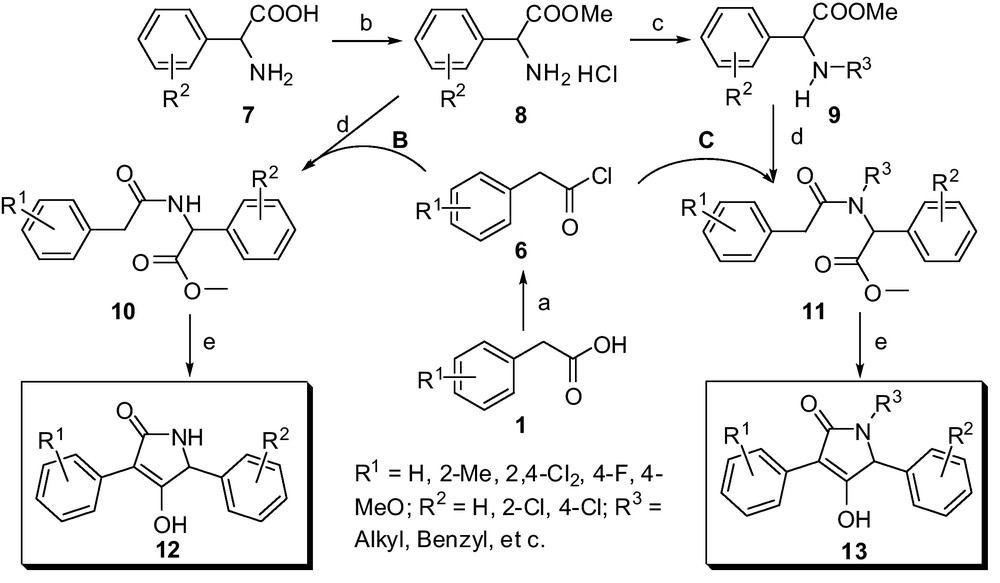
Method B and C. Representative synthetic outline for multi-substituted lactam derivatives based on phenylglycine. Reaction conditions: a. SOCl2, Toluene, reflux 4-5 h; b. SOCl2, MeOH, r.t. to 50 °C; c. Halides, MeCN, K2CO3, r.t.; d. CH2Cl2, Et3N, ice-bath to r.t.; e. tBuOK, THF, reflux for 10 to 15 h.
Having efficient access to butylene lactam derivatives 12 and 13, we could envisage another convenient method for preparation of multi-substituted butylene lactam compounds from easily available aromatic amines. As outlined in Scheme 3, a synthetic route for multi-substituted butylene lactam compounds 17 was described, these were conveniently obtained via three steps including N-alkylation, acylation and intramolecular heterocyclization reactions.

Method D. Synthesis of multi-substituted lactam derivatives 17. Reaction conditions: a: SOCl2, reflux 4 to 5 h; b: α-Halides, MeCN, Et3N, r.t. to 30–40 °C; c: CH2Cl2, Et3N, ice-bath to r.t.; d: tBuOK, THF, reflux for 8 to 12 h.
The various aromatic amines 14 were selected as starting materials, which were conveniently converted to the N-substituted amino-acid ester 15 via N-alkylation reactions under ambient conditions. Subsequently, the acylation and intramolecular heterocyclization reactions are processed with the same as aforementioned procedures with good yields (Table 2). Based on the representative data as shown in Table 2, the electron-donating substituents (4-iPr) and electron-withdrawing groups (4-Br) lead to obviously different reactive activity; the latter need longer reaction time and relatively lower yields. The cooperative effect for N,N-disubstituents of the reactant 16 may be the key factor for the different reactivity. Compound 16c containing 4-isopropyl group has the relatively stable amide bond, which lead to the higher reactivity for the adjacent alpha-carbon of carbonyl group. Generally, these procedures that exploit very easily available raw materials, which will have the widely applied prospects to construct multi-substituted lactam derivatives.
Synthesis of lactam derivatives 17a.
| Entry | 16 | 17 | Substituents | 16b Yield (%) | 17c Yield (%) | ||
| Alkyl | R1 | R2 | |||||
| 1 | 16a | 17a | Me | H | H | 75 | 70 |
| 2 | 16b | 17b | Me | H | 4-Br | 70 | 63 |
| 3 | 16c | 17c | Me | H | 4-iPr | 87 | 77 |
a Representative process for these series derivatives.
b Yields were based on LC-MS analysis.
c Yields refer to isolated products.
Mechanicistically, the one-pot process leading to the formation of butenolides 5 is most probably involving a transesterification and a Dieckmann condensation as the two main steps (Scheme 4), with subsequent acidification to give target compounds.
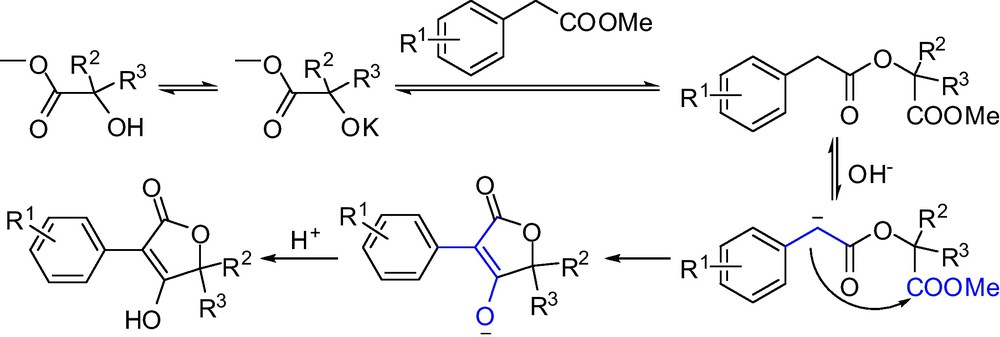
The mechanism of one-pot heterocyclization to provide butenolides.
For the formation of butylene lactams, the intramolecular heterocyclization reaction is very likely initiated by a nucleophilic attack of the α-methyl carbon to the carbonyl carbon of the ester leading to an intermediate, with subsequent keto-enol tautomerism leading to the formation of target compounds. The possible mechanism of ring-closure reaction formation target compounds is shown in Scheme 5.
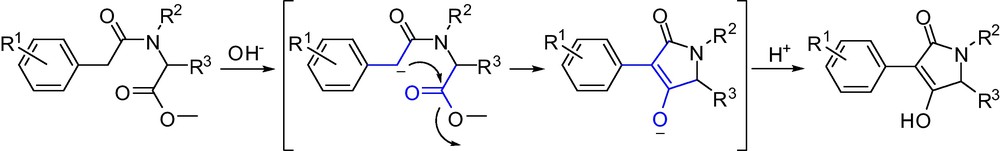
The mechanism of intramolecular heterocyclization reaction to afford lactam.
3 Conclusions
In conclusion, we have described a molecular diversity-oriented convenient process for the assembly of multi-substituted butenolides and lactam derivatives that are important building blocks in the fields of medicine and agrochemicals. In the above-described experimental conditions, the heterocyclization reaction reached completion with a very simple disposal and high yield compared to the reported similar cyclization methods, and a number of functional groups are tolerated by our reaction conditions, including electron-withdrawing and electron-donating groups. Thus, our process represents a versatile access to multi-substituted butenolides and lactam scaffolds using readily available reagents and is a useful tool for the further synthesis of biologically active molecules. The further preparation of other related heterocycles by similar methods is currently underway.
4 Experimental
4.1 Instrumentation and chemicals
All melting points (m.p.) were obtained using a digital model X-5 apparatus and are uncorrected. IR spectra were recorded on a Thermo Nicolet FT-IR Avatar 330 instrument. NMR spectra were recorded on Brucker spectrometer apparatus (1H at 400 MHz or 600 MHz and 13C at 100 MHz with CDCl3 or DMSO-d6 as the solvent). Chemical shifts are reported as δ values (parts per million) relative to internal tetramethylsilane. Coupling constants nJ are reported in Hertz (Hz). Standard abbreviations indicating multiplicity are used as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quadruplet, m = multiplet and br = broad. Mass spectra were performed on a MicroMass Quattro micro™ API instrument. Elemental analysis was performed on a Vario EL III elemental analysis instrument. Analytical thin-layer chromatography (TLC) was carried out on precoated plates, and spots were visualized with ultraviolet light. All chemicals or reagents were purchased from standard commercial supplies. Anhydrous CH2Cl2, CH3OH and THF were prepared by standard methods [22]. All other solvents and reagents were analytical grade and used directly without purification.
4.2 General synthetic procedure for multi-substituted butenolides derivatives 5
The multi-substituted butenolides 5 were prepared following the modified one-pot cyclization methods [23]. The typical procedure is as follows: to a solution of substituted phenylacetate (10 mmol, 1 equiv) and α-hydroxy acids esters (12 mmol, 1.2 equiv) in anhydrous THF was added a solution of potassium tert-butoxide 2.46 g (22 mmol, 2.2 equiv) in THF. The suspension obtained was then refluxed under nitrogen overnight. After cooling to ambient temperature, the reaction mixture was acidified with 5% HCl aqueous to pH 2-3, and then was extracted three times with ethyl acetate. The combined organic layers were then washed with water and dried over Na2SO4. After filtration and concentration under vacuum, the residue was taken up into little ethyl acetate and filtered in vacuo to give the corresponding multi-substituted butenolides 5 as powders. The physicochemical properties and spectral data for representative compounds are as follows:
4.2.1 4-Hydroxy-3-(4-methoxyphenyl)-1-oxaspiro[4.5]dec-3-en-2-one 5a
White powder, yield 75%, m.p. 240.3–241.5 °C (245 °C Lit. [21]); IR (KBr pellet) νmax 833, 969, 1180, 1248, 1388, 1610, 1646, 1686, 2634, 2941, 3437 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 12.19 (bs, 1H, OH), 7.81 (d, J = 7.8 Hz, 2H, Ph-H), 6.94 (d, J = 8.4 Hz, 2H, Ph-H), 3.75 (s, 3H, CH3O), 2.03-1.17 (m, 10H); 13C NMR (100 MHz, DMSO-d6): δ = 178.97, 171.80, 158.40, 128.84, 123.52, 114.14, 97.52, 81.50, 55.69, 32.76, 24.59, 22.34.
4.2.2 3-(4-Fluorophenyl)-4-hydroxy-5-phenylfuran-2(5H)-one 5b
Light yellow powder, yield 75%, m.p. 173.4–175.0 °C; IR (KBr pellet) νmax 697, 834, 1023, 1162, 1381, 1512, 1638, 1707, 2635, 2941, 3408 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 12.60 (bs, 1H, OH), 8.07–8.04 (m, 2H, Ph-H), 7.44-7.18 (m, 7H, Ph-H), 5.75 (s, 1H, CH-O); ESI-MS: calcd for C16H11FO3 m/z 270.1; found, 269.4 (M)+. Anal. Calcd for C16H11FO3: C, 71.11; H, 4.10. Found: C, 71.24; H, 4.02.
4.2.3 3-(2,4-Dichlorophenyl)-4-hydroxy-5-phenylfuran-2(5H)-one 5c
Light yellow powder, yield 78%, m.p. 162.5–163.4 °C; IR (KBr pellet) νmax 698, 996, 1062, 1133, 1260, 1483, 1645, 1713, 2941, 3402 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 12.80 (bs, 1H, OH), 7.73 (s, 1H, Ph-H), 7.52–7.42 (m, 7H, Ph-H), 6.00 (s, 1H, CH-O); ESI-MS: calcd for C16H10Cl2O3 m/z 320.0; found, 319.3 (M+). Anal. Calcd for C16H10Cl2O3: C, 59.84; H, 3.14. Found: C, 59.98; H, 3.07.
4.2.4 4-Hydroxy-3-(4-methoxyphenyl)-5-phenylfuran-2(5H)-one 5d
White powder, yield 71%, m.p. 190.5–192.1 °C; IR (KBr pellet) νmax 696, 835, 1023, 1163, 1248, 1383, 1517, 1609, 1639, 1697, 2645, 2967, 3441 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.88 (d, J = 9.0 Hz, 2H, Ph-H), 7.44 (s, 3H, Ph-H), 7.37 (d, J = 6.0 Hz, 2H, Ph-H), 6.98 (d, J = 8.4 Hz, 2H, Ph-H), 5.88 (s, 1H, CH-O), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6): δ = 174.56, 173.01, 158.58, 136.00, 130.04, 129.52, 128.78, 128.74, 127.33, 123.35, 114.27, 99.12, 78.92, 55.74; ESI-MS: calcd for C17H14O4 m/z 282.1; found, 283.4 (M + H)+. Anal. Calcd for C17H14O4: C, 72.33; H, 5.00. Found: C, 72.51; H, 4.94.
4.3 General synthetic procedure for intermediates substituted 2-phenylacetyl chloride 6
Appropriate substituted phenylacetic acid 1 (10 mmol), 10 mL of toluene and thionyl chloride (5 mL) were placed in a dried round-bottomed flask containing a magnetic stirrer bar and stirred at about 60 °C for 3-6 h. Then the superfluous thionyl chloride was removed by distillation in vacuo at room temperature, and the residue was dissolved in dry CH2Cl2 and the solvent removed as before; this procedure was repeated twice to give intermediates 6, which were used for the next reaction without further purification.
4.4 General synthetic procedure for various amino-acid ester hydrochlorides 8
A suspension of the amino-acid (10 mmol) in absolute methanol (30 mL) was cooled to 0 °C, and thionyl chloride (25 mmol) was added dropwise over 20 min. The cold bath was removed, and the homogeneous solution was heated at reflux for 4 to 6 h. The solvent was removed in vacuo, the resulting residue was dissolved in water (50 mL), and the aqueous solution was extracted with ethyl acetate and then evaporated in vacuo to the corresponding amino-acid ester hydrochlorides 8.
4.5 General synthetic procedure for N-substituted amino-acid ester 9
To a solution of the corresponding amino-acid ester hydrochlorides (10 mmol) in anhydrous acetonitrile (30 mL) was added K2CO3 (25 mmol) in batches over 20 min. The cold bath was removed, and various halides added to the aforementioned reaction mixture. The heterogeneous solution was stirred at ambient condition for 5 to 8 h. The solvent was removed in vacuo, the resulting residue was dissolved in water (50 mL), and the aqueous solution was extracted with ethyl acetate and then evaporated in vacuo to the corresponding N-substituted amino-acid ester 9, which were used for the next reaction without further purification.
4.6 General procedure for synthesis of intermediates 10 and 11
The typical process for synthesis of phenylacetamide derivatives 10 is as follows: The freshly formed acylchloride 6 (2.2 mmol) dissolved in dry dichloromethane was added dropwise to a solution of the substituted amino-acid ester hydrochloride (2 mmol), and Et3N (5 mmol) in dry dichloromethane under ice-bath. The resultant solution was stirred at room temperature for 2 to 5 h, which was detected by TLC. Then the mixture was washed to neutral with water and dried via anhydrous Na2SO4. After filtration and concentration, the organic residues were used for the next cyclization reaction without further purification. The N-substituted phenylacetamide derivatives 11 were conveniently obtained with the similar procedure as that for compounds 10.
4.7 General synthetic procedure for target compounds lactam derivatives 12 and 13
To a solution of phenylacetamide derivatives 10 (10 mmol, 1 equiv) in anhydrous THF was added in batches with potassium tert-butoxide (12 mmol, 1.2 equiv). The suspension mixture obtained was then refluxed under nitrogen overnight. After cooling to ambient temperature, the reaction mixture was acidified with 5% HCl aqueous to pH 2-3, and then extracted three times with ethyl acetate. The combined organic layers were then washed with water and dried over Na2SO4. After filtration and concentration under vacuum, the residue was taken up with ethyl acetate and filtered in vacuo to give the corresponding multi-substituted lactam derivatives 12. The N-substituted lactam derivatives 13 were prepared according to the above-described method. The physicochemical properties and spectral data for representative compounds are as follows:
4.7.1 5-Benzyl-4-hydroxy-3-phenyl-1H-pyrrol-2(5H)-one 12a
White floccule, yield 80%, m.p. 212.0–214.0 °C; IR (KBr pellet) νmax 692, 784, 1090, 1274, 1304, 1458, 1610, 1637, 2926, 3434 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.35 (bs, 1H, OH), 7.78 (d, J = 7.9 Hz, 2H, Ph-H), 7.49 (s, 1H, NH), 7.27–7.09 (m, 8H, Ph-H), 4.23 (s, 1H, CH-N), 3.12 (q, J = 6.0 Hz, 1H, CH2), 2.94 (q, J = 6.0 Hz, 1H, CH2); ESI-MS: calcd for C17H15NO2 m/z 265.1; found, 266.6 (M + H)+. Anal. Calcd for C17H15NO2: C, 76.96; H, 5.70; N, 5.28. Found: C, 76.90; H, 5.75; N, 5.34.
4.7.2 5-Benzyl-3-(2,4-dichlorophenyl)-4-hydroxy-1H-pyrrol-2(5H)-one 12b
White powder, yield 70%, m.p. 113.5–115.1 °C; IR (KBr pellet) νmax 702, 751, 823, 1235, 1364, 1475, 1561, 1643, 1737, 2950, 3268, 3449 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.60 (d, J = 8.0 Hz, 1H, Ph-H), 7.53 (s, 1H, NH), 7.34–7.22 (m, 7H, Ph-H), 4.54-4.48 (m, 1H, CH-N), 3.06 (q, J = 5.4 Hz, 1H, CH2), 2.90 (q, J = 9.5 Hz, 1H, CH2); ESI-MS: calcd for C17H13Cl2NO2 m/z 333.0; found, 334.6 (M + H)+. Anal. Calcd for C17H13Cl2NO2: C, 61.10; H, 3.92; N, 4.19. Found: C, 61.17; H, 3.86; N, 4.24.
4.7.3 5-Benzyl-3-(4-fluorophenyl)-4-hydroxy-1H-pyrrol-2(5H)-one 12c
Light yellow powder, yield 75%, m.p. 203.0–205.0 °C; IR (KBr pellet) νmax 733, 840, 1084, 1237, 1297, 1318, 1511, 1610, 1655, 2926, 3068, 3438 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.40 (bs, 1H, OH), 7.86 (q, 4J = 5.8 Hz, J = 8.9 Hz, 2H, Ph-H), 7.52 (s, 1H, NH), 7.25–7.06 (m, 7H, Ph-H), 4.23 (t, J = 4.7 Hz, 1H, CH-N), 3.12 (q, J = 4.0 Hz, 1H, CH2), 2.94 (q, J = 4.0 Hz, 1H, CH2); 13C NMR (100 MHz, DMSO-d6): δ = 173.12, 170.20, 161.96, 136.84, 130.29, 129.40, 129.33, 128.47, 127.02, 115.17, 114.96, 103.96, 56.39, 37.73; ESI-MS: calcd for C17H14FNO2 m/z 283.1; found, 284.7 (M + H)+. Anal. Calcd for C17H14FNO2: C, 72.07; H, 4.98; N, 4.94. Found: C, 72.01; H, 5.04; N, 4.88.
4.7.4 5-Benzyl-4-hydroxy-3-(4-methoxyphenyl)-1H-pyrrol-2(5H)-one 12d
Yellow powder, yield 77%, m.p. 194.2–195.6 °C; IR (KBr pellet) νmax 703, 736, 840, 1176, 1251, 1287, 1513, 1611, 1653, 2935, 3057, 3432 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.20 (bs, 1H, OH), 7.73 (d, J = 8.8 Hz, 2H, Ph-H), 7.47 (s, 1H, NH), 7.23–7.15 (m, 5H, Ph-H), 6.84 (d, J = 8.8 Hz, 2H, Ph-H), 4.22 (t, J = 4.4 Hz, 1H, CH-N), 3.72 (s, 3H, CH3O), 3.11 (q, 2J = 13.7 Hz, J = 3.7 Hz, 1H, CH2), 2.93 (q, 2J = 13.7 Hz, J = 5.8 Hz, 1H, CH2); ESI-MS: calcd for C18H17NO3 m/z 295.1; found, 296.8 (M + H)+. Anal. Calcd for C18H17NO3: C, 73.20; H, 5.80; N, 4.74. Found: C, 73.13; H, 5.86; N, 4.81.
4.7.5 4-Hydroxy-3,5-diphenyl-1H-pyrrol-2(5H)-one 12e
White powder, yield 78%, m.p. 191.4–192.5 °C; IR (KBr pellet) νmax 692, 703, 775, 918, 1197, 1313, 1382, 1498, 1593, 1659, 2578, 2943, 3069, 3411 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.25 (bs, 1H, OH), 8.01 (s, 1H, NH), 7.95 (d, J = 7.2 Hz, 2H, Ph-H), 7.41–7.30 (m, 7H, Ph-H), 7.19 (t, J = 7.4 Hz, 1H, Ph-H), 5.03 (s, 1H, CH-N); ESI-MS: calcd for C16H13NO2 m/z 251.1; found, 252.6 (M + H)+. Anal. Calcd for C16H13NO2: C, 76.48; H, 5.21; N, 5.57. Found: C, 76.39; H, 5.27; N, 5.51.
4.7.6 3-(2,4-Dichlorophenyl)-4-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one 12f
Light yellow powder, yield 68%, m.p. 195.2–196.7 °C; IR (KBr pellet) νmax 697, 748, 828, 1099, 1191, 1322, 1382, 1480, 1581, 1626, 1659, 2515, 2920, 3290, 3437 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.23 (bs, 1H, OH), 8.05 (s, 1H, NH), 7.63 (s, 1H, Ph-H), 7.44–7.32 (m, 7H, Ph-H), 5.07 (s, 1H, CH-N); ESI-MS: calcd for C16H11Cl2NO2 m/z 319.0; found, 320.6 (M + H)+. Anal. Calcd for C16H11Cl2NO2: C, 60.02; H, 3.46; N, 4.37. Found: C, 60.13; H, 3.56; N, 4.32.
4.7.7 3-(4-Fluorophenyl)-4-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one 12 g
Light yellow powder, yield 64%, m.p. 179.2–180.5 °C; IR (KBr pellet) νmax 576, 701, 784, 843, 1221, 1382, 1507, 1599, 1634, 1682, 2571, 2918, 3414 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.35 (bs, 1H, OH), 8.05–8.01 (m, 3H, NH and Ph-H), 7.41–7.29 (m, 5H, Ph-H), 7.17 (t, J = 9.0 Hz, 2H, Ph-H), 5.03 (s, 1H, CH-N); ESI-MS: calcd for C16H12FNO2 m/z 269.1; found, 270.6 (M + H)+. Anal. Calcd for C16H12FNO2: C, 71.37; H, 4.49; N, 5.20. Found: C, 71.32; H, 4.56; N, 5.13.
4.7.8 3-(2,4-Dichlorophenyl)-4-hydroxy-5-(2-(methylthio)ethyl)-1H-pyrrol-2(5H)-one 12 h
Light yellow powder, yield 65%, m.p. 185.0–186.0 °C; IR (KBr pellet) νmax 671, 778, 825, 1099, 1200, 1316, 1409, 1483, 1584, 1623, 1659, 2607, 2912, 3259, 3441 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.28 (bs, 1H, OH), 7.74 (s, 1H, NH), 7.61 (d, J = 2.1 Hz, 1H, Ph-H), 7.40 (q, 4J = 2.1 Hz, J = 8.3 Hz, 1H, Ph-H), 7.27 (d, J = 8.3 Hz, 1H, Ph-H), 4.10 (t, J = 4.6 Hz, 1H, CH-N), 2.12–1.69 (m, 7H, CH2CH2SCH3); ESI-MS: calcd for C13H13Cl2NO2S m/z 317.0; found, 318.6 (M + H)+. Anal. Calcd for C13H13Cl2NO2S: C, 49.07; H, 4.12; N, 4.40. Found: C, 49.15; H, 4.06; N, 4.44.
4.7.9 7-Hydroxy-6-phenyl-2,3-dihydro-1H-pyrrolizin-5(7aH)-one 12i
White powder, yield 68%, m.p. 212.0–214.0 °C; IR (KBr pellet) νmax 632, 692, 787, 918, 941, 1230, 1304, 1340, 1572, 1650, 2560, 2896, 2975, 3443 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.92 (bs, 1H, OH), 7.20-7.99 (m, 5H, Ph-H), 4.19 (t, J = 6.4 Hz, 1H, CH-N), 1.40–3.09 (m, 6H, CH2CH2CH2); ESI-MS: calcd for C13H13NO2 m/z 215.1; found, 216.6 (M + H)+. Anal. Calcd for C13H13NO2: C, 72.54; H, 6.09; N, 6.51. Found: C, 72.46; H, 6.15; N, 6.43.
4.7.10 6-(2,4-Dichlorophenyl)-7-hydroxy-2,3-dihydro-1H-pyrrolizin-5(7aH)-one 12j
White powder, yield 74%, m.p. 226.0–228.0 °C; IR (KBr pellet) νmax 638, 772, 820, 870, 1099, 1221, 1367, 1340, 1578, 1608, 1653, 2685, 2887, 2949, 3441 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.78 (bs, 1H, OH), 7.60 (s, 1H, Ph-H), 7.39 (q, 4J = 1.6 Hz, 3J = 8.4 Hz, 1H, Ph-H), 7.26 (d, J = 8.4 Hz, 1H, Ph-H), 4.16 (t, J = 7.9 Hz, 1H, CH-N), 4.15-3.00 (m, 2H, CH2), 2.17–1.36 (m, 4H, CH2); ESI-MS: calcd for C13H11Cl2NO2 m/z 283.0; found, 284.6 (M + H)+. Anal. Calcd for C13H11Cl2NO2: C, 54.95; H, 3.90; N, 4.93. Found: C, 55.03; H, 3.84; N, 4.85.
4.7.11 6-(4-Fluorophenyl)-7-hydroxy-2,3-dihydro-1H-pyrrolizin-5(7aH)-one 12k
White powder, yield 76%, m.p. 228.6–230.2 °C; IR (KBr pellet) νmax 644, 775, 834, 941, 1162, 1233, 1299, 1352, 1388, 1418, 1516, 1576, 1653, 2631, 2896, 2971, 3442 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.90 (bs, 1H, OH), 8.01 (q, 2H, Ph-H), 7.15 (t, J = 9.0 Hz, 2H, Ph-H), 4.14 (q, J = 8.4 Hz, 1H, CH-N), 3.41–3.02 (m, 2H, CH2), 2.20–1.33 (m, 4H, CH2); ESI-MS: calcd for C13H12FNO2 m/z 233.1; found, 234.6 (M + H)+. Anal. Calcd for C13H12FNO2: C, 66.94; H, 5.19; N, 6.01. Found: C, 66.87; H, 5.12; N, 6.07.
4.7.12 7-Hydroxy-6-o-tolyl-2,3-dihydro-1H-pyrrolizin-5(7aH)-one 13a
White powder, yield 62%, m.p. 165.3–167.7 °C; IR (KBr pellet) νmax 632, 724, 745, 781, 942, 1227, 1364, 1400, 1590, 1611, 2673, 2890, 2965, 3437 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.36 (bs, 1H, OH), 7.20–7.08 (m, 4H, Ph-H), 4.15 (q, J = 6.7 Hz, 1H, CH-N), 3.40–3.02 (m, 2H, CH2), 2.19–1.38 (m, 7H, CH2 and CH3); 13C NMR (100 MHz, DMSO-d6): δ = 177.69, 172.54, 137.80, 131.45, 131.34, 130.22, 127.64, 125.69, 106.79, 63.93, 43.90, 29.28, 28.57, 20.41; ESI-MS: calcd for C14H15NO2 m/z 229.1; found, 230.6 (M + H)+. Anal. Calcd for C14H15NO2: C, 73.34; H, 6.59; N, 6.11. Found: C, 73.27; H, 6.66; N, 6.03.
4.7.13 7-Hydroxy-6-(4-methoxyphenyl)-2,3-dihydro-1H-pyrrolizin-5(7aH)-one 13b
White solid, yield 72%, m.p. 218.7–220.2 °C; IR (KBr pellet) νmax 638, 769, 837, 1019, 1177, 1245, 1382, 1432, 1516, 1596, 1650, 2676, 2896, 2959, 3458 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.68 (bs, 1H, OH), 7.88 (d, J = 8.2 Hz, 2H, Ph-H), 6.89 (d, J = 8.2 Hz, 2H, Ph-H), 4.12 (t, J = 7.1 Hz, 1H, CH-N), 3.42–3.35 (m, 1H, CH2), 3.33 (s, 3H, CH3O), 3.02–3.00 (m, 1H, CH2), 2.21–2.14 (m, 1H, CH2), 2.08-2.03 (m, 2H, CH2), 1.38-1.31 (m, 1H, CH2); ESI-MS: calcd for C14H15NO3 m/z 245.1; found, 246.7 (M + H)+. Anal. Calcd for C14H15NO3: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.47; H, 6.23; N, 5.65.
4.7.14 1-(4-Bromobenzyl)-4-hydroxy-3,5-diphenyl-1H-pyrrol-2(5H)-one 13c
White powder, yield 74%, m.p. 179.3–181.0 °C; IR (KBr pellet) νmax 641, 692, 739, 769, 909, 1013, 1072, 1188, 1310, 1376, 1489, 1599, 1623, 1638, 2673, 2920, 3446 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.74 (d, J = 7.8 Hz, 2H, Ph-H), 7.47–7.14 (m, 10H, Ph-H), 6.95 (d, J = 7.8 Hz, 2H, Ph-H), 5.03 (d, 2J = 15.6 Hz, 1H, CH2-N), 4.67 (s, 1H, CH-N), 3.51 (d, 2J = 15.0 Hz, 1H, CH2-N); 13C NMR (100 MHz, DMSO-d6): δ = 171.36, 169.70, 138.08, 135.91, 132.48, 132.03, 130.44, 129.56, 129.40, 129.14, 128.51, 127.94, 126.75, 120.77, 103.99, 63.22, 42.85; ESI-MS: calcd for C23H18BrNO2 m/z 419.1; found, 420.4 (M + H)+.
4.8 General procedure for synthesis of intermediates 15
The various aromatic amines 14 were selected as starting materials, which were conveniently converted to N-substituted amino-acid ester 15 via N-alkylation reactions under ambient conditions. The typical processes are as follows: To a solution of various aromatic amines (10 mmol) and Et3N (15 mmol) in anhydrous acetonitrile (20 mL) was added α-halides (12 mmol) dropwise over 30 min. The mixture was stirred at ambient condition, which was monitored by TLC. Then the reaction was worked-up with normal procedure to access to the corresponding N-substituted amino-acid ester 15, which were used for the next reaction without further purification.
4.9 General synthetic procedure for the multi-substituted lactam derivatives 17
The N-substituted lactam derivatives 17 were prepared according to the aforementioned procedures via acylation and intramolecular heterocyclization reactions. The physicochemical properties and spectral data for representative compounds are as follows:
4.9.1 4-Hydroxy-5-methyl-1,3-diphenyl-1H-pyrrol-2(5H)-one 17a
White powder, yield 70%, m.p. 229.1–231.2 °C; IR (KBr pellet) νmax 701, 778, 887, 1105, 1266, 1389, 1495, 1590, 1629, 1656, 2676, 2949, 3447 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.94 (d, J = 7.8 Hz, 2H, Ph-H), 7.57 (d, J = 7.8 Hz, 2H, Ph-H), 7.40–7.35 (m, 4H, Ph-H), 7.21 (t, J = 6.9 Hz, 1H, Ph-H), 7.12 (t, J = 7.5 Hz, 1H, Ph-H), 4.84 (d, J = 6.6 Hz, 1H, CH-N), 1.33 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ = 171.98, 169.35, 138.05, 132.39, 129.41, 128.47, 128.07, 126.68, 124.17, 122.38, 103.49, 54.85, 17.36; ESI-MS: calcd for C17H15NO2 m/z 265.1; found, 266.4 (M + H)+.
4.9.2 1-(4-Bromophenyl)-4-hydroxy-5-methyl-3-phenyl-1H-pyrrol-2(5H)-one 17b
White solid, yield 63%, m.p. 240.2–242.1 °C; IR (KBr pellet) νmax 596, 701, 778, 810, 897, 1010, 1099, 1194, 1394, 1486, 1590, 1626, 1673, 2697, 2976, 3438 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.93 (d, J = 7.8 Hz, 2H, Ph-H), 7.57 (s, 4H, Ph-H), 7.36 (t, J = 7.4 Hz, 2H, Ph-H), 7.22 (t, J = 7.2 Hz, 1H, Ph-H), 4.85 (d, J = 6.0 Hz, 1H, CH-N), 1.34 (d, J = 6.6 Hz, 3H, CH3); ESI-MS: calcd for C17H14BrNO2 m/z 343.0; found, 344.4 (M + H)+.
4.9.3 4-Hydroxy-1-(4-isopropylphenyl)-5-methyl-3-phenyl-1H-pyrrol-2(5H)-one 17c
White solid, yield 77%, m.p. 229.0–230.1 °C; IR (KBr pellet) νmax 703, 781, 837, 906, 1102, 1200, 1284, 1391, 1516, 1596, 1620, 1676, 2695, 2955, 3447 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.95 (s, 2H, Ph-H), 7.46 (d, J = 8 Hz, 2H, Ph-H), 7.35 (t, J = 7.4 Hz, 2H, Ph-H), 7.26-7.20 (m, 3H, Ph-H), 4.78 (d, J = 6.6 Hz, 1H, CH-N), 2.88 (t, J = 6.9 Hz, 1H, CH(CH3)2), 1.32 (d, J = 6.6 Hz, 3H, CH3), 1.22 (d, J = 7.2 Hz, 6H, CH(CH3)2); ESI-MS: calcd for C20H21NO2 m/z 307.2; found, 308.5 (M + H)+.
Acknowledgements
Financial support from the Special Fund for Agro-Scientific Research in the Public Interest (200903052) and the National Natural Science Foundation of China (31000867) is kindly acknowledged. The authors also would like to thank many colleagues of the Hubei Biopesticide Engineering Research Center and the Hubei Academy of Agricultural Sciences for valuable advice and assistance.