

1 Introduction
Cyclodextrins (CD) are macrocyclic oligomers of α–D-glucose [1]. They are shaped like truncated cones with primary and secondary hydroxyl groups crowning the narrower rim and wider rim, respectively. Three species of CDs are the most widely used [2]. They have rings comprising from six to eight glucose units: α-CD (six units), β-CD (seven units), γ-CD (eight units). Because CDs have a hydrophilic exterior and a hydrophobic cavity of appropriate dimension, they can bind with various guest molecules to form inclusion complexes in aqueous solution [3]. The β-CD is the most widely used in the CD family, but it presents a solubility of only 1.5 g/mL at 25 °C in water, which is a limiting factor for its use. In order to enhance the solubility of the β-CD in aqueous media, totally and partially methylated β-CD derivatives have been synthetesized [4]. Methylated CDs have attracted considerable attention due to their solubility both in water [5] and in organic solvents [4]. Moreover, inclusion complexes of methylated CDs are usually more stable than the corresponding complexes of unmodified CDs [5]. Some β-cyclodextrin derivatives (heptakis-2, 6-di-O-methyl-β-CD [Dimeb]), amorphous randomly substituted methyl-β-CD (RAMEB) and semi-crystalline methyl-β-CD (Crysmeb) were investigated and compared with those of natural (α-, β- and γ-) CD [6–10]. These modified CDs have been widely used, for example, as enzyme mimics, supramolecular receptors and chiral selectors in separation science and technology [11].
Doxycycline (DOX), a semi-synthetic derivative of oxytetracycline, exists in two tautomeric forms (keto and enol) in which the keto-enol equilibrium in aqueous media is in favor of the enol form [12]. It is a potent antibacterial drug commonly used as DOX hyclate (DOX-h). As other tetracyclines, it is remarkably tissue-irritating when injected. DOX-h spectrum may be useful to treat important bacterial infections in calves, such as pneumonias, skin and soft tissue infections, genital tract and gastrointestinal tract bacterial diseases such as salmonelosis and colibacilosis [13].
The formation of the DOX:Crysmeb complex allows one to reduce irritation and enhance the absorption rate.
Recently, the DOX:Crysmeb complex was investigated using REOSY NMR technique [12]. Thus, a stoichiometry of 1:1 had been determined for the complexation process and the authors revealed that the DOX molecule was included in the Crysmeb cavity by its aromatic ring [12]. Unfortunately, this investigation had not allowed the authors to give us a clear picture on this inclusion complex. In order to provide complementary information on the DOX: Crysmeb complex and a better understanding of the interactions of such inclusion processes of the CDs, and to complete experiment results, we undertaken a theoretical study using quantum mechanics calculations.
In the present work, we will determine the energy, the interactions and the geometry of Crysmeb complexation with DOX using semi-empirical PM6 and ONIOM [B3LYP/6-31G(d):RHF/3-21G*] methods [14–22].
2 Computational method
In the present investigation, all the calculations were carried out with MOPAC 20091 and the Gaussian 03 programs [23]. The molecular structures of DOX and Crysmeb were built by the Chem-Office package2. The geometries of keto and enol tautomeric forms of DOX were optimized using the density functional theory (DFT) method with B3LYP hybrid functional at the 6-31G* level and the Crysmeb molecule was optimized at the Hartree-Fock (HF) level with the basis set 3-21G* (Fig. 1). For the construction of DOX:Crysmeb complex, the glycosidic oxygen atoms of β-CD were placed onto the XY plane and their centre was defined as the origin of the coordinate system. Bond 1–2 of DOX is coincident with the Z axis and the relative position between DOX and Crysmeb was measured by the X coordinate of carbon atom 1 of DOX (Fig. 2). The inclusion complexation was emulated by entering the guest molecule from the wide end of Crysmeb and then letting it pass through the Crysmeb in several steps. In each step, the geometry of the complex was fully optimized without any restriction using the PM6 method. This procedure of complexation was adapted for the keto and enol forms of DOX. DOX molecule was initially located at Z coordinates of –8 Å and was moved through the β-CD cavity along the Z axis to –2 Å with a step of 0.5 Å. In order to find an even more stable structure of the complex, each DOX molecule was rotated around the C1-C2 bond of DOX that coincide with the Z-axis, by 20° intervals from 0° to 360°. The complexation energy Ecomplexation was defined as the difference between the sum of energy of individual host and guest molecules and the energy of inclusion complex. In PM6 calculations, we replaced the potential energy term by the heat of formation in Eq. (1).
| (1) |

Top view of (a) doxycycline (DOX) (b) Crysmeb; a: RB3LYP (6-31G*) optimized doxycycline enol form (right) and keto form (left); b: 2-O-methyl-βCD (Crysmeb) and glycopyranose unit.

The relative position between the doxycycline (DOX) and the Crysmeb.
After that, ONIOM [RB3LYP/6-31G(d):RHF/3-21G*] calculations were carried out with the energy minimum of tautomeric forms. In ONIOM method, the high level RB3LYP/6-31G* is carried out on DOX and the low level RHF/3-21G* on CRYSMEB. The integrated energy for the two-layered ONIOM approach is defined as:
| E (ONIOM2) = E (High, Model) + E (Low, Real)–E (Low, Model) | (2) |
3 Results and discussion
The results of the inclusion process in the cavity of Crysmeb for both enol and keto forms of DOX obtained by PM6 method are summarized in Fig. 3 and Table 1. The Fig. 3 illustrates the stabilization energy variation of the inclusion processes for both enol and keto forms of DOX into Crysmeb at different distances. The minimum energy for the enol form is located at –4 Å but for the keto form is located at –5 Å. All the energy values obtained by the two latter methods are calibrated by inclusion of zero-point energy (ZPE) correction.
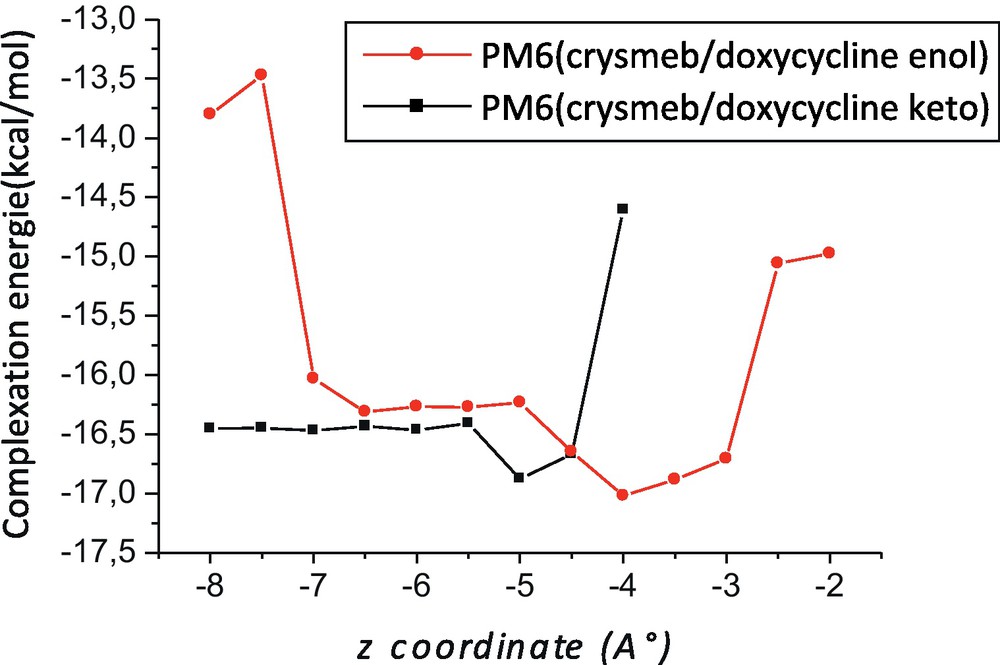
Complexation energies of the inclusion complexation of doxycycline (DOX) into Crysmeb at different positions (Z) and for both forms; a: enol; b: keto.
Heat of formation, binding and complexation energy obtained by PM6.
| Crysmeb/doxycycline enol | Crysmeb/doxycycline keto | ΔE = Eketo – EEnol | |
| ΔHf | –1868.16 | –1867.57 | 0.59a 0.63b |
| Ebinding | –24.07 | –23.66 | 0.41a 0.44b |
| Ecomplexation | –16.87 | –17.02 | 0.14a 0.17b |
a Difference of complexation energy between the two forms ΔE = Eenol – E1keto.
b ΔE zeo-point energy correction.
The negative binding energy changes upon complexation clearly demonstrate that Crysmeb can form stable complexes with DOX (enol and keto form) but favor the enol form of 0.44 and 0.17 kcal/mol, respectively, in agreement with experiment observations [12].
The structures with minimum energy obtained from ONIOM calculations for the enol and keto forms of DOX are shown in Fig. 4. DOX tend to bond strongly with Crysmeb by penetrating its cavity by the wider and the more accessible face. However, the cavity of Crysmeb is just wide enough to allow full penetration for enol and keto forms of DOX. Moreover, the DOX molecule is too large to fit entirely in the cavity and the effect of the steric barrier becomes large after natural β-CD was modified, which causes difficulties to the DOX molecule to penetrate into the Crysmeb cavity. In fact, an inspection of the geometry of the two complexes shows that the aromatic ring of DOX entered fully into the cavity of Crysmeb, while the methyl-6 group remains on the rim of Crysmeb on the one hand and the rest of molecule keep outside the hydrophilic exterior of the cavity on the other hand. This obvious configuration is due certainly to the presence of CO, CONH2 and N(CH3)3 groups which add charged sites on the DOX and make it more hydrophilic.

ONIOM [RB3LYP/6-31G(d):RHF/3-21G*]; a: Crysmeb/doxycycline enol; b: Crysmeb/doxycycline Keto.
According to the results highlighted in Table 2, the enol form was found significantly more favorable than the keto form by the binding and the complexation energy difference of 20.71 and 6.27 kcal/mol, respectively, with ONIOM (B3LYP/6-31G(d):RHF/3-21G*) (Table 2). These results included the scaled ZPE correction.
Binding and complexation energy ONIOM (RB3LYP/6-31G(d):RHF/3-21G*).
| Complex (Enol) | Complex (keto) | ΔE = Eketo – EEnol | |
| Ebinding | –51.06 | –29.49 | 21.57a 20.71b |
| EComplexation | –22.66 | –17.52 | 5.14a 6.27b |
a Difference of complexation energy between the two orientations ΔE = Eenol – Eketo.
b ΔE zero-point energy (ZPE) correction.
We can see in Fig. 4 the formation of several hydrogen bonds in the favorable structure (enol form) obtained with two layers ONIOM [B3LYP/6-31G (d):RHF/3-21G*] method. Thus, in the case of where DOX is regarded as an H-bond donor, two conventional H-bonds were expected. The first one between the oxygen atom (O53) and the hydrogen atom (H218) of H218-O197 bond, separated by 1.8 Å and with angle equal to 153.8°. The second one between the oxygen atom (O73) and the hydrogen atom (H214) of H214-O195 bond, separated by 1.7 Å and with an angle equal to 150° (Fig. 4).
These conventional H-bonds are assisted by three weak H-bonds namely which can play a significant role in the stability of inclusion complexes. The oxygen atom O54 can establish two weak H-bonds. The first one with the hydrogen atom (H219) of H219-C197 bond separated of 2.2 Å and with an angle equal to 151.7°. The second one is expected with the hydrogen atom (H222) of H222-C200 bond separated by 2.4 Å and an angle equal to 151.8°. The oxygen atom O45 is expected to give weak hydrogen bond with a hydrogen atom (H205) of H205-C177 bond separated by 2.4 Å and an angle equal to 152.0° (Fig. 5). However, in the case where DOX is regarded as acceptor group, only one H-bond was established between the oxygen atom (O197) and the hydrogen atom (H140) of H140-O59 bond separated by 1.7 Å and an angle equal to 169.4° (Fig. 4).
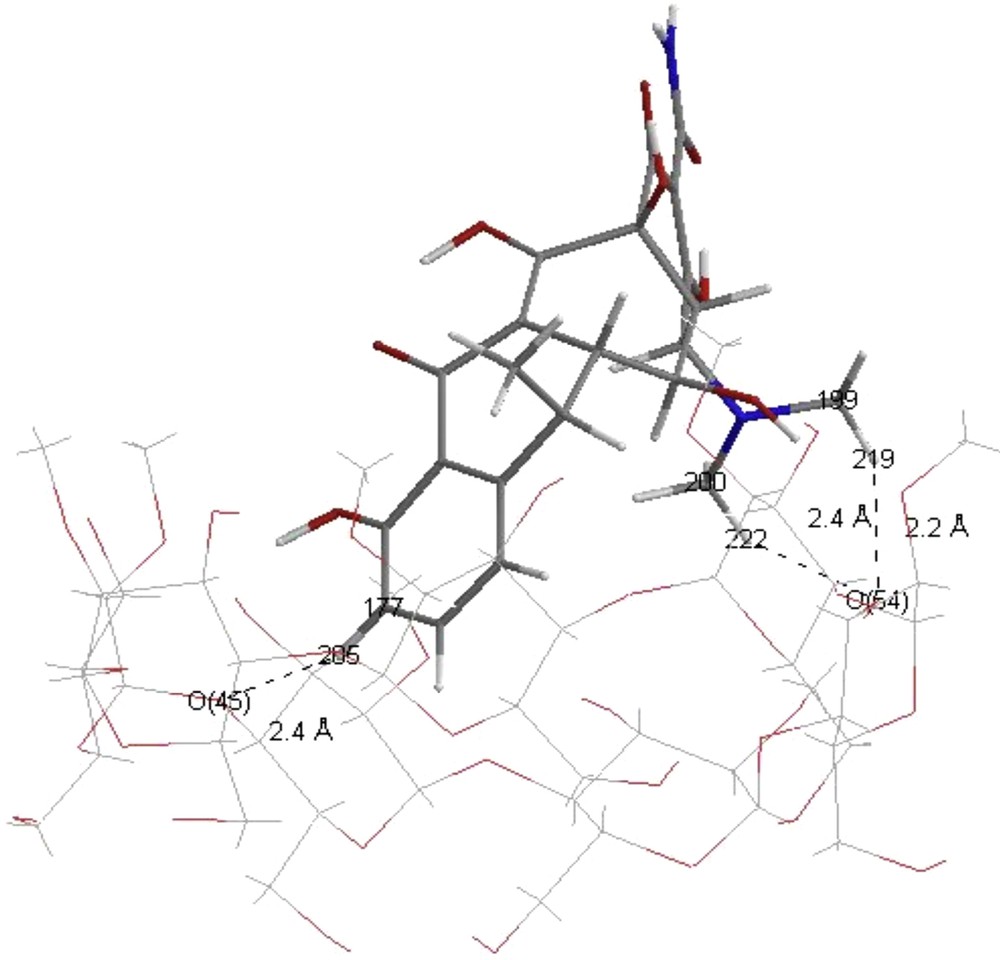
Weak H-bonds in Crysmeb/doxycycline enol: ONIOM [RB3LYP/6-31G(d):RHF/3-21G*].
4 Natural Bond Orbital (NBO) analysis
The formation of a hydrogen bonded complex implies that a certain amount of electronic charge is transferred from the proton accepter to the proton donor [24]. Delocalization effects can be identified from the presence of diagonal elements of the Fock matrix in the NBO basis. The strengths of these delocalization interactions E(2) are estimated by second order perturbation theory [25].
The stabilization energies E(2) calculated using RB3LYP/6-31G(d) single point methods and the geometric parameters of the established H-bond in the inclusion complex are presented in Table 3. Indeed, as it can be seen, significant interaction energies are obtained for the expected hydrogen bonds. Approximately, it was stated that conventional hydrogen bond variable to 1 to 10 kcal/mol while the weak H-bond namely for which energies vary between 0.5 and 2 kcal/mol [26]. Indeed, we can consider the obtained values (Table 3) of H-bond energies as acceptable except the conventional H-bond which is underestimated (1.32 kcal/mol) by comparison with the values obtained for weak H-bond. Similar results were obtained in the previous study [27,28].
The electron donor and accepter orbital, the corresponding E(2) hydrogen bonds interactions energies, distances and angles obtained with RB3LYP/6-31G(d).
| Crysmeb/doxycycline enol | Natural Bond Orbital (NBO) donor | Natural Bond Orbital acceptor | E(2) (Kcal/mol) |
| Crysmeb proton acceptor and doxycycline donor | |||
| LP(1) O53 | BD*O197-H218 | 5.72 (1.8 Å, 153.8°) | |
| LP(1) O73 | BD*O195-H214 | 1.32 (1.7 Å, 150.0°) | |
| LP(1) O45 | BD*C177-H205 | 1.76 (2.5 Å, 151.6°) | |
| LP(1) O54 | BD*C199-H219 | 1.96 (2.2 Å, 151.7°) | |
| LP(2) O54 | BD*C200-H222 | 1.70 (2.4 Å, 145.3°) | |
| Doxycycline proton acceptor and Crysmeb donor | |||
| LP(1) O197 | BD*O59-H140 | 2.32 (1.7 Å, 169.7°) |
We can notice an interesting interaction that was detected by RMN only in the enol form [12]. This interaction occurs between hydrogen atoms H108 of DOX and H208 of cavity of Crysmeb. As it was expected, NBO calculation confirms the existence of this Van der Waals interaction only in the enol form and was estimated at 1.37 kcal/mol. This involved an approaching of the two hydrogen atoms; the distance of H-bond is 1.9 Å in the enol form while this distance became equal to 2.8 Å in the keto form (Fig. 4).
Intramolecular hydrogen bonding can be also a significant factor in determining the preference in DOX:Crysmeb complex. First of all, it is important to know that several intramolecular H-bonds can occur in DOX molecule and its inclusion in the CD cavity can allow their disappearance or maybe the appearance of the new ones. Firstly, the oxygen atom (O192) as well as in the free DOX-keto or DOX-enol forms, establish a very strong intramolecular H-bond with hydrogen atom (H212) of O191-H212 bond, and with stabilization energy E(2) equal to 9.20 kcal/mol and 9.35 kcal/mol, respectively. However, these values of energy become lower in the DOX-keto and DOX-enol inclusion complex, 6.44 kcal/mol and 5.89 kcal/mol, respectively. In both free form of the DOX intramolecular H-bond was detected with hydrogen atom (H204) of H204-O176 bond the oxygen atom (O192) and is predicted more favorable with enol form with E(2) equal to 7.37 kcal/mol. Secondly, the oxygen atom (O175) establish intramolecular H-bond with hydrogen atom (H203) of N176-H203 bond only in the free DOX-keto and DOX-enol forms with stabilization energy E(2) equal to 7.73 and 7.86 kcal/mol, respectively.
Finally, intramolecular H-bond was detected just in DOX-keto form between the oxygen atom (O193) and hydrogen atom (H214) of O182-H214 bond with E(2) equal to 5.13 kcal/mol which had disappeared completely in its inclusion complex; and yet, in the DOX-enol form, the same oxygen atom O193 establish intramolecular H-bond with hydrogen atom (H213) of O194-H213 bond with E(2) equal to 6.32 kcal/mol which was accompanied by a decrease of the E(2) value in its inclusion complex to attain 5.57 kcal/mol (Fig. 6, Table 4).

The intramolecular hydrogen bond of the doxycycline (DOX); a: doxycycline enol in complex; a1: doxycycline enol free; b: doxycycline keto incomplex; b1: doxycycline keto free.
Intramolecular hydrogen bond in free doxycycline and in their inclusion complex.
| Enol | Keto | ||||
| Natural Bond Orbital (NBO) Donor | Natural Bond Orbital (NBO) acceptor | Incl-comp | Free | Incl-comp | Free |
| LP O192 | BD*O191-H212 | 5.89 (1.9 Å) | 9.20(1.5 Å) | 6.44 (1.9 Å) | 9.35(1.5 Å) |
| LP O192 | BD*N176-H204 | 7.37 (1.9 Å) | – | 7.14 (1.9 Å) | – |
| LP O175 | BD*N176-H203 | – | 7.73(1.9 Å) | – | 7.86(1.9 Å) |
| LP O193 | BD*O194-H213 | 5.57 (1.6 Å) | 6.32(1.6 Å) | – | – |
| LP O193 | BD*O182-H214 | – | – | 5.13 (1.7 Å) | – |
5 Conclusion
The results obtained by employing ONIOM2 method provide important insights into the geometry and the interactions between DOX and Crysmeb molecules. These results clearly show that the enol form is more favorable than the keto form, in agreement with mass and NMR spectroscopy observations. The geometry of the most stable complex shows that the aromatic ring is included inside in the hydrophobic cavity of β-CD while the rest of DOX molecule keeps outside. The intermolecular interactions such as hydrogen bonding (conventional and weak H-bond) and hydrophobic interactions are the key players in stabilizing energetically of the most favorable structure of DOX:Crysmeb complex.
Acknowledgement
This paper was supported by the Algerian Ministry of Higher Education and Scientific Research and General Direction of Scientific and technological research as a part of projects CNEPRU projects (No: E01520080026 and No: D01520100004) and PNR (8/u24/4814). We acknowledge the department of chemistry at Guelma University in which this work was performed.
1 Stewart J.J.P., MOPAC2009 version 9.034W, web: http://openMOPAC.net.
2 Version 6.0, Cambridge software.