

1 Introduction
The growing concern for the environment demands development of green and financially viable processes for the synthesis of organic compounds. Synthetic chemists in both academia and industry are constantly challenged to consider more environmentally benign methods for generation of the target molecules. In this sense, utilizing “safer solvents” and to “design for energy efficiency” can be considered two key principles of green chemistry, relevant to synthetic chemists. From this point of view, non-classical methods, microwave-assisted synthesis, ultrasonic irradiation, use of ionic liquids and supercritical fluids find applications as tempting methods to achieve these goals [1–3]. The use of ultrasound activation in organic transformation is now well known to enhance the reaction rate, selectivity and yield of the reaction [4,5]. The driving energy is provided through cavitation, the formation and collapse of bubbles, which liberates considerable energy in very short times. A literature survey reveals that, compared to traditional methods, this approach is faster and facile and is more suitable as far as consideration of green chemistry is concerned [6,7].
On the other hand, the use of non-aqueous room temperature ionic liquids (ILs) as environmental benign solvents in organic transformations has gained considerable importance due to their negligible vapour pressure, reusability and high thermal stability [8,9]. Apart from this, some ILs possess inherent Lewis/Brønsted acidity, which can promote and catalyze organic transformations of commercial importance in excellent yields. The use of ionic liquids as reaction media may offer a convenient solution to both the solvent emission and catalytic recycling problem with high efficacy [10–12]. The use of ultrasound in combination with ILs, which have no vapour pressure, may force the less volatile substrates to undergo the cavitational activation by changing the characteristics of cavitation and push the reaction in the forward direction very quickly. The synergy of ultrasound and ionic liquid in catalyst-free methodologies for the synthesis of heterocyclic compounds has attracted much interest because of their less hazardous nature, reduced energy consumption and increased product selectivity [13,14].
In the families of heterocyclic compounds, xanthene core and its derivatives serve as an important class of compounds, as it is present in natural products with broad biological activities [15–18]. Most notably among them, xanthenedione constitutes a structural unit in a number of natural products, and has a wide range of therapeutic and pharmacological properties [19–21]. Several functionalized derivatives of xanthene possess antiplasmodial [22], antiviral [23], antibacterial [24] and anti-inflammatory [25] activities. Besides, these heterocyclic molecules have been widely used as luminescent dyes [26,27], sensitizers in photodynamic therapy [28–31], in laser technology [32,33] as well as pH sensitive fluorescent materials [34,35]. Attempts to synthesize xanthenediones over various catalysts such as sulfuric acid or hydrochloric acid [36], InCl3/ionic liquid [37], Fe+3 montmorilonite [38], amberlyst-15 [39], FeCl3/[bmim][BF4] [40], p-dodecylbenzenesulphonic acid [41], sulphamic acid [42], HClO4·SiO2 [43], trimethylsilyl chloride (TMSCl) [44] have been reported. Most of the reported methods have used expensive reagents, hazardous organic solvents, longer reaction time and tedious workup. Hence, there is scope for further innovation towards contemporary reaction with easy isolation of product, reusability of catalyst, perhaps with minimal or no waste.
As a part of our continuing studies in developing efficient catalyst-free synthetic methodologies, using ultrasound and ionic liquid in organic preparations [45], herein, we report a general method for the synthesis of 1,8-dioxo-octahydroxanthenes promoted by synergistic effect of ultrasonic irradiation and ionic liquid at ambient temperature without any added catalyst.
2 Results and discussion
To find out the optimum reaction condition, for the sonochemical synthesis of titled compounds (3a–u), the condensation reaction between benzaldehyde (1) and dimedone (2) was chosen as a model reaction under different reaction conditions (Scheme 1). The obtained results are recorded in Table 1. It is clear from the results that 1,8-dioxo-octahydroxanthene can be synthesized with high yield and purity at ambient temperature using 200 mg IL in 1 mL methanol as the co-solvent without any added catalyst, in 50 min. To examine the scope and general applicability of the methodology, a broad range of structurally diverse aromatic aldehydes (aromatic aldehydes having electron withdrawing and electron donating groups as well as hetero aromatic aldehydes) was condensed with dimedone under the found optimum reaction condition. All the reactions were monitored by thin layer chromatography (TLC) and taken to completion. The time taken for the completion of each conversion, aldehyde and the isolated yields are summarized in Table 2. It can be observed that all the aldehydes have reacted in short reaction times (45–85 min) under these mild conditions to afford the xanthenes in very good to excellent isolated yields. The process was tolerated well by all the aldehydes irrespective of their nature of substituents. Methanol was added as co-solvent to solubilize dimedone at ambient temperature. In absence of MeOH, the reaction proceeded with poor yield (Entry 4, Table 1), and the TLC of reaction mixture also indicated the presence of unreacted dimedone. The reaction, in absence of IL, did not result in the formation of product even after ultrasonication for 180 min (Entry 1, Table 1). All the synthesized xanthenes were homogeneous on TLC and pure enough for the further practical purpose. However, all the compounds were crystallized from hot ethanol. All the synthesized compounds were characterized by melting point, 1H NMR, 13C NMR and DEPT-135 spectral techniques. Additional confirmation for the structures is also obtained by IR and mass spectrometric studies for some representative samples. All the data were in agreement with the cited literature.
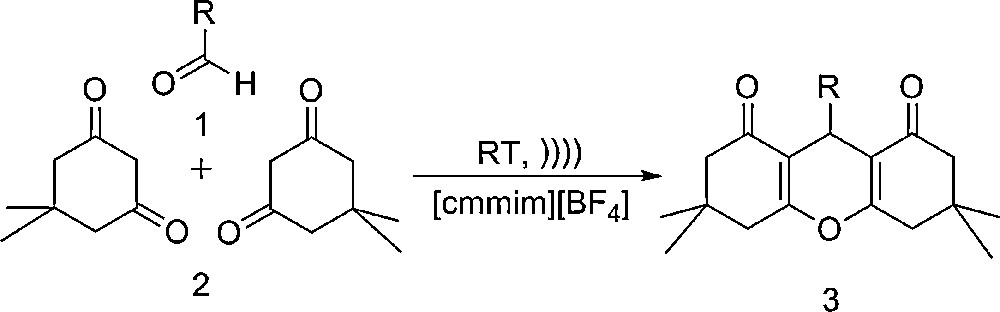
Catalyst-free one-pot synthesis of 1,8-dioxo-octahydroxanthene under ultrasonic irradiations in ionic liquid at ambient temperature.
Optimization of reaction condition for the synthesis of 1,8-dioxo-octahydroxanthene.
| Entry | IL/co-solvent | Reaction condition | Time (min)a | % Yield |
| 1 | No IL/2 mL MeOH | RT,))) | 180 | 0 |
| 2 | [cmmim][BF4]/1 mL MeOH | RT,))) | 50 | 87 |
| 3 | [cmmim][BF4]/1 mL MeOH | 50 °C,))) | 50 | 89 |
| 4 | [cmmim][BF4]/no co-solvent | RT,))) | 180 | 28b |
| 5 | [cmmim][BF4]/1 mL MeOH | RT, stirring | 180 | Intermediate |
a All the reactions were run till completion as indicated by TLC.
b Dimedone remains sparingly soluble under this condition.
Catalyst-free synthesis of 1,8-dioxo-octahydroxanthenes under ultrasonication at ambient temperature using variously substituted aromatic aldehydes.
| Compound | R | Time (min) | Yield (%)a | M.P. (°C) | |
| Found | Reported | ||||
| 3a | C6H5 | 50 | 87 | 198–200 | 199–201 [37] |
| 3b | 4-O2NC6H4 | 60 | 85 | 222–224 | 222–224 [37] |
| 3c | 2-ClC6H4 | 65 | 86 | 226–228 | 225–227 [37] |
| 3d | 3-ClC6H4 | 55 | 82 | 184–186 | 183–185 [47] |
| 3e | 4-ClC6H4 | 55 | 81 | 230–232 | 226–228 [47] |
| 3f | 2-O2NC6H4 | 70 | 83 | 258–260 | 258–262 [48] |
| 3g | 3-O2NC6H4 | 60 | 87 | 166–168 | 165–166 [47] |
| 3h | 4-H3CC6H4 | 55 | 78 | 216–218 | 215–216 [47] |
| 3i | 4-MeOC6H4 | 45 | 85 | 240–242 | 242–243 [47] |
| 3j | 4-HOC6H4 | 80 | 79 | 246–248 | 247–248 [47] |
| 3k | 4-HO,3-MeOC6H3 | 85 | 81 | 226–228 | 225–227 [47] |
| 3l | 2-BrC6H4 | 45 | 84 | 226–228 | 226–229 [47] |
| 3m | 3-BrC6H4 | 55 | 86 | 192–194 | 190–192 [50] |
| 3n | 4-FC6H4 | 45 | 88 | 224–226 | 226–227 [47] |
| 3o | 2,5-(MeO)2C6H3 | 40 | 82 | 172–174 | 174–175 [47] |
| 3p | 3,4-(MeO)2C6H3 | 55 | 86 | 174–176 | 175–176 [47] |
| 3q | 3,4,5-(MeO)3C6H2 | 55 | 83 | 186–188 | 187–189 [50] |
| 3r | 2-HOC6H4 | 50 | 84 | 202–204 | 202–205 [49] |
| 3s | 2-Furyl | 30 | 89 | 60–62 | 62–64 [50] |
| 3t | 2-Thienyl | 45 | 92 | 164–166 | 164–165 [47] |
| 3u | 2-Pyridyl | 50 | 90 | 204–206 | 203–205 [47] |
a Yield after crystallization.
To investigate the role of ultrasound, the model reaction was carried out under simple stirring at ambient temperature (Entry 5, Table 1). No formation of product 3a was observed even after 180 min of continuous stirring. The absence of ultrasonic irradiation brings the reaction to stop at the intermediate stage, the 1H NMR spectrum of the intermediate is shown on Fig. 1 for the perusal. The NMR spectrum corresponds well with the uncyclised intermediate involving two enols and two ketons, since the H on methylene bridge is a singlet. The integration for hydroxyl proton is one instead of two; this may probably be due to a broad signal and exchange with residual HDO/D2O. The NMR data is in good agreement with an earlier work by Singh et al. [46]. The increase in the rate and selectivity of the reaction under ultrasonication appears to be due to the occurrence of cavitation. Ultrasound travelled through alternating compressions and rarefactions in the transmitting medium along the direction of wave propagation [47,48]. Small cavities or gas filled micro bubbles are formed due to a large negative pressure on liquid in the rarefaction section of the wave. An acoustic cavitation, which includes nucleation, growth and collapse of the cavity, is responsible for the chemical and physical effects of ultrasound [49]. The energy released involved with cavitation is best explained in terms of the “hot spot” approach. Each cavitation bubble acts as a localized microreactor and its violent collapse generates the energy for chemical effects. Due to negligible vapour pressure and strong cohesive forces, it is difficult to induce cavitation in an ionic liquid. Therefore, in such a medium, the cavitational bubble will contain the vapour of reaction partners and co-solvent. The ionic liquid shell surrounds it, forces it to go through strong cavitational effects.
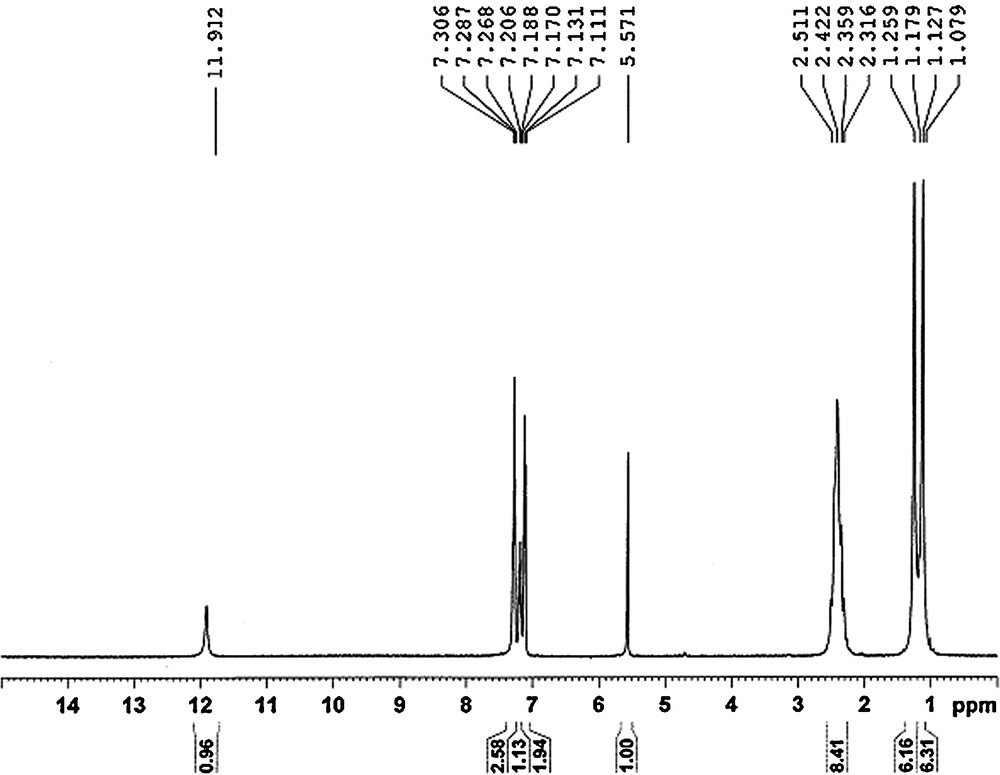
1H NMR spectrum of uncyclised intermediate.
The IL promotes the reactions due to its inherent Brønsted acidity at ambient temperature. In our previous study [45], we demonstrated that hydrogen bonding can be formed between carboxylic proton of IL and carbonyl oxygen of aldehyde during the reaction. Accordingly, the same hydrogen bonding can also be formed between the carboxylic proton of the IL and the carbonyl oxygen of dimedone. The formation of a hydrogen bond between the IL and the substrate leads them to activation. Based on this investigation, we suggest the plausible mechanistic pathway for this reaction (Scheme 2).
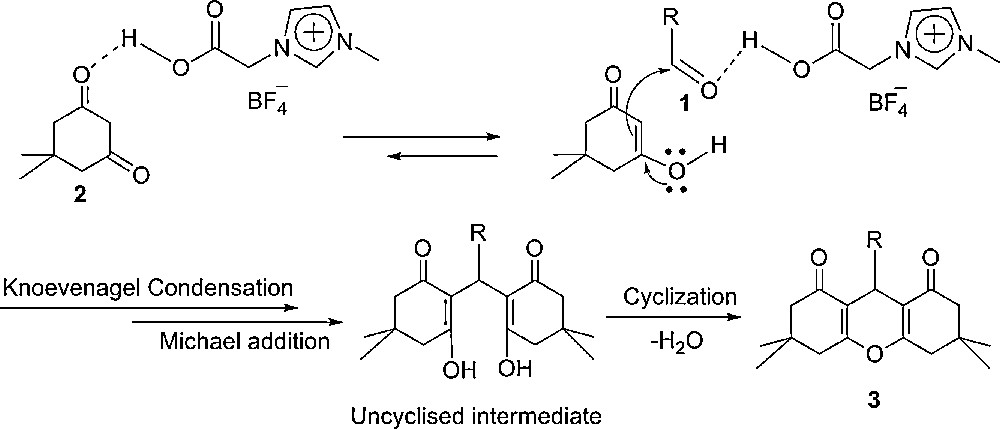
Plausible mechanistic pathway for the synthesis of 1,8-dioxo-octahydroxanthene.
To investigate the efficiency of the recycled ionic liquid, we ran six successive cycles of the model reaction at ambient temperature under the optimum reaction conditions using recycled IL from the previous run. The ionic liquid was found to be effective for at least six reaction cycles with prominent retention in its activity. The obtained results are plotted in Fig. 2.

Recyclability of ionic liquid in the model reaction between benzaldehyde and dimedone.
3 Conclusion
The sonochemical reaction provided an improved practical alternative to access functionalized xanthenes in excellent yields. The beneficial features of the reaction, such as mild conditions, absence of added catalyst and reusability of the ionic liquid gives it an advantage over the conventional acid/base catalyzed reaction.
4 Experimental
4.1 General
All chemicals were of research grade and were used as obtained from Sigma-Aldrich, Alfa-Aesar or SDFCL. The IL was prepared under ultrasonic irradiation, according to the method reported earlier [45]. The melting points were determined in capillary tubes using heavy paraffin liquid in Thiele tube. Melting points are uncorrected and are compared with the reported literature values. The reaction progress and purity of products were determined by TLC silica gel plates (Merck 60 F254). IR Spectra were recorded on a Shimadzu FT-IR-S8401 and FT-IR-8400 spectrophotometer using KBr, mass spectra on AB APPLIED BIOSYSTEMS IMDS SCIEX. API-2000 LC/MS/MS spectrometer. The 1H NMR (400 MHz), 13C NMR (100 MHz) and DEPT-135 spectra were recorded on BRUKER AVANCE 400 MHz instrument using CDCl3 as the solvent and TMS as an internal standard. All the reactions were carried out in an ultrasonic cleaning bath (D-compact ultrasonic cleaner) at 50 kHz frequency and 250 W power output. The reactions were carried out in a round-bottomed flask of 25 mL capacity suspended at the center of the cleaning bath, 5 cm below the surface of the liquid. The temperature of water bath was controlled at 30 °C by addition or removal of water from the bath.
4.2 General procedure for the synthesis of 1,8-dioxo-octahydroxanthenes
A mixture of 5,5-dimethyl-1,3-cyclohexanedione (2 mmol), aldehyde (1 mmol) and [cmmim][BF4] (200 mg) was charged into a 25 mL flask. The mixture was stirred gently with a spatula for a few seconds to ensure homogeneous mixing of reactants with ionic liquid. One milliliter of methanol was added to it. Then, the reaction mixture was subjected to ultrasonic irradiation at 30 °C. After completion of reaction (as indicated by TLC), the reaction mixture was poured onto crushed ice (∼20 gm) and stirred well. The solid separated was washed with ice-cold water (∼ 4 × 5 mL), and then crystallized from hot ethanol to afford pure 1,8-dioxo-octahydroxanthenes. The aqueous filtrate was subjected to vacuo at 80 °C under reduced pressure (10 mmHg) for 4 h to leave behind the IL in near complete recovery, pure enough for the next run.
4.3 Spectral data of selected compounds
4.3.1 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-phenyl-2H-xanthene-1,8(5H,9H)-dione (compound 3a)
1H NMR (400 MHz, CDCl3): δ 7.29 (m, 2H), 7.23 (m, 2H), 7.11 (m, 1H), 4.77 (s, 1H), 2.48 (s, 4H), 2.21 (dd, 4H, J = 16.4 Hz), 1.12 (s, 6H), 1.01 (s, 6H). 13C NMR: δ 27.34, 29.26, 31.84, 32.19, 40.90, 50.76, 115.70, 126.36, 128.04, 128.38, 144.10, 162.22, 196.32. DEPT-135: up peaks: δ 27.34, 29.26, 31.84, 126.36, 128.04, 128.38. Down peaks: δ 40.89, 50.75. IR (KBr): 3030, 2955, 1662, 1624, 1361, 1197, 1163 cm−1.m/z (ESI): 351.0 [M + H+].
4.3.2 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-(4-nitrophenyl)-2H-xanthene-1,8(5H,9H)-dione (compound 3b)
1H NMR (400 MHz, CDCl3): δ 8.10 (m, 2H), 7.49 (m, 2H), 4.84 (s, 1H), 2.51 (s, 4H), 2.22 (dd, 4H, J = 16.4 Hz), 1.13 (s, 6H), 1.00 (s, 6H). 13C NMR: δ 27.28, 29.23, 32.22, 32.37, 40.58, 50.61, 114.54, 123.42, 129.36, 146.50, 151.51, 162.95, 196.23. DEPT-135: up peaks: δ 27.27, 29.23, 32.37, 123.42, 129.36. Down peaks: δ 40.84, 50.60. IR (KBr): 3020, 2956, 1660, 1516, 1361, 1199, 1004 cm−1.m/z (ESI): 396.0 [M + H+].
4.3.3 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-(4-methylphenyl)-2H-xanthene-1,8(5H,9H)-dione (compound 3h)
1H NMR (400 MHz, CDCl3): δ 7.02 (m, 2H), 6.82 (m, 2H), 4.92 (s, 1H), 2.51 (s, 4H), 2.29 (s, 3H), 2.19 (dd, 4H, J = 16.3 Hz), 1.09 (s, 6H), 1.00 (s, 6H). 13C NMR: δ 21.20, 27.23, 29.24, 31.95, 32.18, 42.06, 50.09, 112.11, 125.23, 129.25, 135.26, 141.50, 162.54, 195.83. DEPT-135: up peaks: δ 21.20, 27.23, 29.24, 32.18, 125.23, 129.25. Down peaks: δ 42.06, 50.09. IR (KBr): 3135, 2954, 1720, 1592, 1378, 1191, 1081 cm−1.
4.3.4 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-(4-hydroxy-3-methoxyphenyl)-2H-xanthene-1,8(5H,9H)-dione (compound 3k)
1H NMR (400 MHz, CDCl3): δ 6.93 (m, 1H), 6.71 (m, 1H), 6.64 (m, 1H), 5.81 (s, 1H), 4.66 (s, 1H), 3.88 (s, 3H), 2.44 (s, 4H), 2.19 (dd, 4H, J = 16.3 Hz), 1.10 (s, 6H), 1.01 (s, 6H). 13C NMR: δ 27.31, 29.15, 31.98, 32.08, 42.36, 51.13, 55.92, 111.13, 113.16, 117.12, 120.54, 132.31, 145.56, 148.23, 162.21, 196.12. DEPT-135: up peaks: δ 27.31, 29.15, 32.08, 55.92, 111.13, 117.12, 120.54. Down peaks: δ 42.36, 51.13. IR (KBr): 3692, 3581, 3155, 2964, 2228, 1645, 1509, 1362, 1164 cm−1.
4.3.5 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-(3-bromophenyl)-2H-xanthene-1,8(5H,9H)-dione (compound 3m)
1H NMR (400 MHz, CDCl3): δ 7.42-6.94 (m, 4H), 4.86 (s, 1H), 2.43 (s, 4H), 2.23 (dd, 4H, J = 16.0 Hz), 1.21 (s, 6H), 1.10 (s, 6H). 13C NMR: δ 27.31, 29.51, 32.06, 32.51, 42.13, 51.24, 112.65, 122.89, 125.23, 127.21, 130.26, 131.86, 141.92, 162.23, 196.46. DEPT-135: up peaks: δ 27.31, 29.51, 32.51, 125.23, 127.21, 130.26, 131.86. Down peaks: δ 42.13, 51.24. IR (KBr): 3020, 2965, 2886, 1594, 1364, 1215 cm−1.
4.3.6 3,4,6,7-tetrahydro-3,3,6,6-tetramethyl-9-(2-thienyl)-2H-xanthene-1,8(5H,9H)-dione (compound 3t)
1H NMR (400 MHz, CDCl3): δ 7.82-7.98 (m, 2H), 6.78 (m, 1H), 4.92 (s, 1H), 2.53 (s, 4H), 2.26 (dd, 4H, J = 16.0 Hz), 1.13 (s, 6H), 1.01 (s, 6H). 13C NMR: δ 27.18, 29.33, 31.02, 31.89, 41.06, 49.63, 113.69, 119.39, 124.01, 132.26, 136.25, 161.84, 196.33. DEPT-135: up peaks: δ 27.18, 29.33, 31.89, 119.39, 124.01, 132.26. Down peaks: δ 41.06, 49.63. IR (KBr): 2955, 2896, 2871, 1659, 1624, 1371, 1360, 1201 cm−1.
Acknowledgements
Authors thank the Head, Department of Chemistry, Sardar Patel University for providing necessary research facilities. A.N. Dadhania also thanks University Grant Commission – New Delhi, India for the meritorious fellowship.
Vous devez vous connecter pour continuer.
S'authentifier