

1 Short overview of the ticlopidine and clopidogrel story
Clopidogrel is an oral antiplatelet agent indicated for the reduction of atherothrombotic events in patients with recent myocardial infarction (MI), recent stroke or established peripheral arterial disease. It was indeed shown to reduce the rate of a combined endpoint of new ischemic stroke (fatal or not), new MI (fatal or not) and other vascular death in these patients. It is also used in combination with aspirin to treat new/worsening chest pain (new heart attack, unstable angina) and to keep blood vessels open and prevent blood clots after certain procedures (such as cardiac stent, coronary bypass graft, balloon angioplasty). Its tolerance is clearly better than that of its predecessor, ticlopidine. Clopidogrel is marketed by Sanofi and Bristol-Myers Squibb under the trade name Plavix. In 2010, it was the second-best selling drug with $ 9.4 billion in global sales but generics are now on the market. A third generation thienopyridine, Prasugrel, was recently approved for the reduction of thrombotic cardiovascular events (including stent thrombosis) in patients with acute coronary syndrome who are to be treated with percutaneous coronary intervention (PCI).
Clopidogrel is a thienopyridine molecule, which is structurally related to ticlopidine (Fig. 1), the first member of this class of antiplatelet agents. The story started in 1972 when my manager, Dr Fernand Eloy, decided to search for new anti-inflammatory drugs related to Tinoridine (Fig. 1), a thienopyridine compound whose anti-inflammatory and analgesic properties were published 2 years previously by the Yoshitomi Company [1]. Exploiting our knowledge in thienopyridine chemistry, we synthesized a number of derivatives in gram quantities [2] to screen them on a wide battery of animal models exploring different physiological systems or mimicking human pathologies. Almost all these tests were performed in vivo or ex-vivo, in the mouse and the rat. It appeared that none of the thienopyridine compounds synthesized had anti-inflammatory or analgesic effects but, fortunately, some of them displayed unexpected antiplatelet and antithrombotic activities after oral administration to rats. It is worth mentioning that it was uncommon, at that time, to search for new antiplatelet agents. The link between platelet aggregation, thrombosis and cardiovascular events was still ignored or disputed by some cardiologists and in actual fact, vascular spasm was considered as the major cause of the clinical complications of atherosclerosis. One of the most active compounds found, ticlopidine, was rapidly selected for development and it was first evaluated in clinical conditions where platelet interaction with artificial surfaces can lead to thrombotic complications, for example in cardiac surgery with extracorporeal circulation, or in patients undergoing hemodialysis. The drug was marketed in France, in 1978, for these restricted clinical indications, under the name Ticlid. Subsequent large clinical trials proved the effectiveness of ticlopidine in other patients at high risk of thrombosis events, notably those with previous transient ischemic attacks and stroke, peripheral arterial disease or ischemic heart disease. The product was then used worldwide, reaching the US market in 1991.
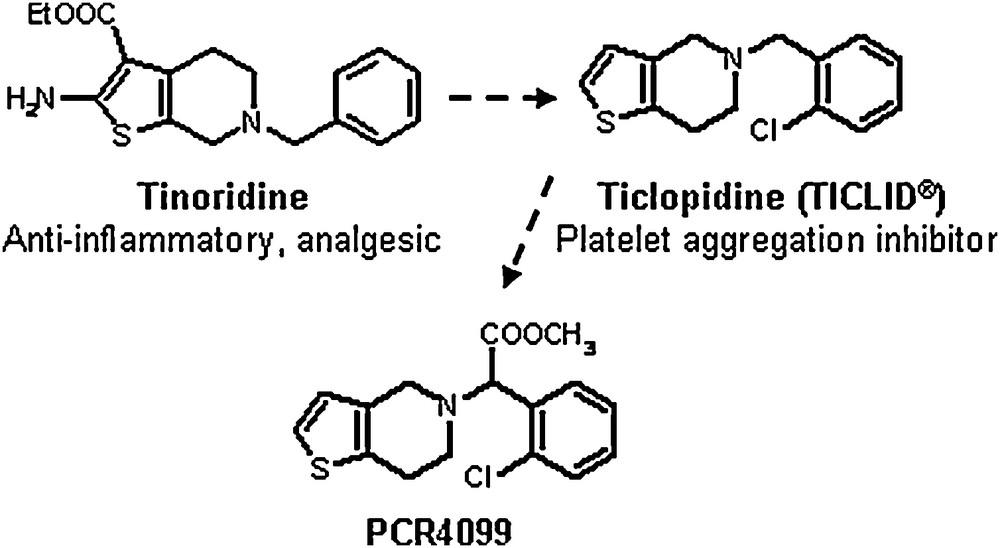
From tinoridine to ticlopidine and PCR4099.
However, as soon as ticlopidine was selected for preclinical development, we continued to synthesize thienopyridine analogues to try to get backup compounds with a better activity/toxicity ratio in animals, which could translate, into a better benefit/risk ratio in humans. This objective became more crucial when, a few months after the launching of ticlopidine in France, it appeared that a few of the patients treated suffered from severe hematological disorders including leucopenia, thrombocytopenia, agranulocytosis and pancytopenia. These life-threatening adverse effects were confirmed and quantified in subsequent large clinical trials and post-marketing pharmacovigilance. Most of them occurred during the first 3 months of treatment and were reversible on discontinuation of therapy but, unfortunately, they cannot be predicted by any identified demographic or clinical characteristics. Therefore, the Health Authorities required that patients treated with ticlopidine must be hematologically and clinically monitored during that 3-month period.
In total, more than a thousand ticlopidine analogues were synthesized and tested in animals for their antiplatelet and antithrombotic effects. Eight of them were developed up to phase 1 studies in healthy volunteers and only the last one, PCR4099 (Fig. 1), proved to be clearly more active and better tolerated than ticlopidine. Further preliminary phase II clinical studies confirmed its powerful antiplatelet effects in patients with previous thrombotic events.
Actually, PCR4099 is a racemic mixture of two enantiomers in equal amounts [3] (Fig. 2). Because we were very concerned by the first reported adverse reactions of ticlopidine in man and because we wanted to develop the best backup possible, we decided to try to separate the two enantiomers to see if, by chance, one of them could have a better activity/toxicity ratio, keeping in mind that it was impossible to reliably reproduce in animals or in in vitro models of human hematopoietic cells the rare ticlopidine hematological effects observed in man. After a number of unsuccessful attempts, we set up a separation method, which could be industrially extrapolated [4]. It appeared that only the dextrogyre (S)-isomer (clopidogrel) had antiplatelet and antithrombotic effects while the inactive (-) - (R)-isomer was clearly less well tolerated in animals. Consequently, we stopped the clinical studies of the racemic PCR4099 and started the preclinical development of clopidogrel, as a hemisulfate salt, in 1987. After 10 years’ development and large clinical studies, the product was launched worldwide in 1998.
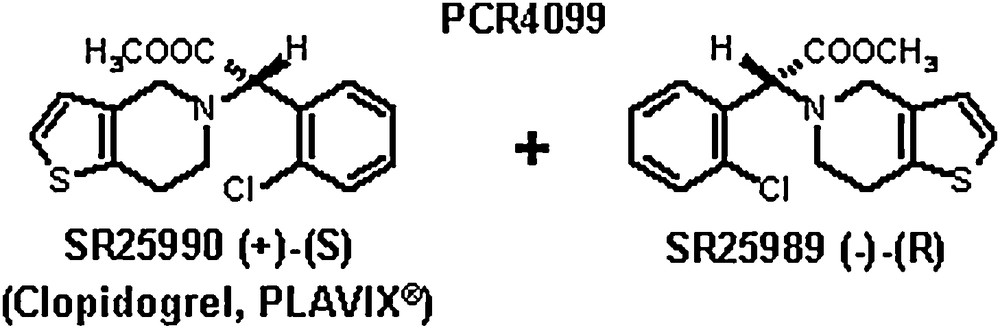
PCR4099 is a racemic mixture: only the dextrogyre (S)-isomer (clopidogrel) has antiplatelet and antithrombotic effects while the inactive levogyre isomer is less well tolerated.
When clopidogrel entered into preclinical development, its mode of action (and that of ticlopidine) was not fully elucidated but it was obvious that it differed from that of other platelet inhibitors such as aspirin, sulfinpyrazone and dipyridamole. At variance with these drugs, it appeared as a powerful inhibitor of ADP-induced platelet aggregation [5]. We were also aware that, due to their different mechanism of action, the combination of clopidogrel and aspirin potentiates their antiplatelet and antithrombotic activity in animals. We also knew that clopidogrel is a prodrug: it is inactive in vitro and it needs a hepatic metabolization to be effective [6]. We were convinced that the active metabolite is chemically and biologically unstable and that it irreversibly affects platelets for the remainder of their lifespan thus explaining its long duration of action. But we did not have knowledge of:
- • the chemical structure of the active metabolite(s);
- • the platelet receptor affected by the active metabolite(s);
- • the enzymes (CYPs450) involved in the formation of this active metabolite.
The active metabolite was isolated and characterized in 2000 [7] (Fig. 3) and its platelet target was identified in 2001 as the P2Y12 receptor of ADP [8] (Fig. 4), soon after its cloning [9]. The enzymes involved in the active metabolization of clopidogrel were progressively identified, the last one (PON-1) being proposed in 2011 [10] but not subsequently confirmed (cf. section 4). In other words, it took more than 30 years after the discovery of ticlopidine and more than 10 years after the synthesis of clopidogrel to elucidate the mechanism of action of these two major antithrombotic drugs.
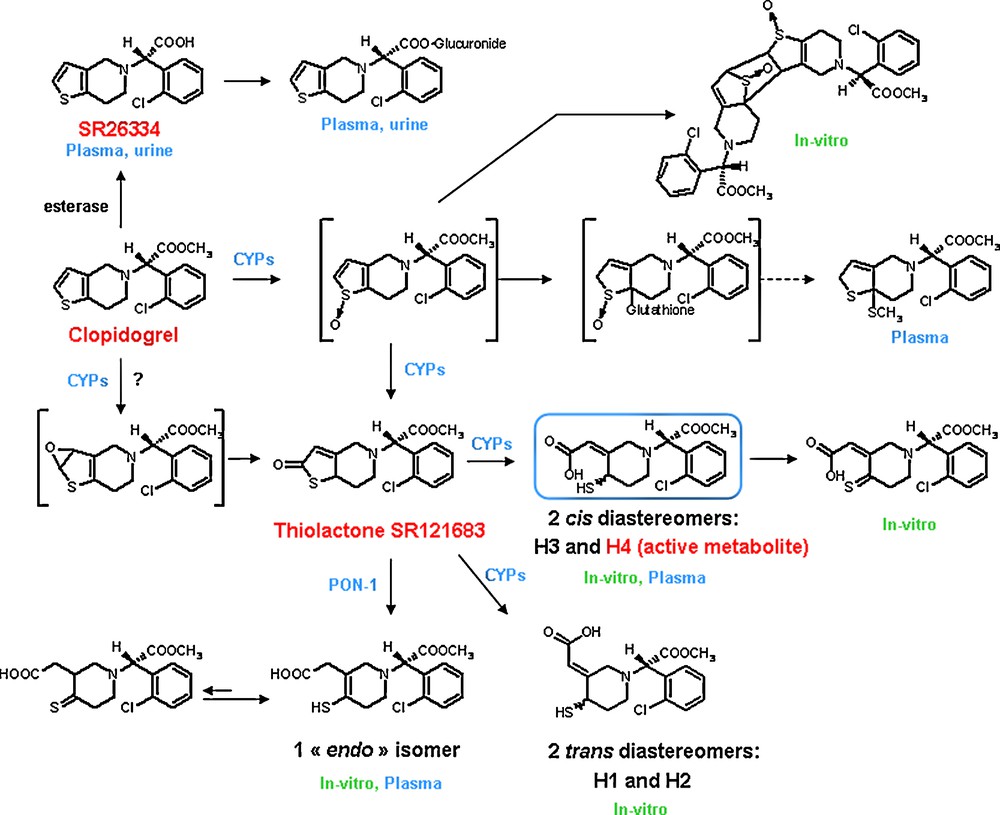
Clopidogrel metabolism in humans (in vitro and in vivo). H4 is the only in vivo metabolite which is active in vitro on platelets.
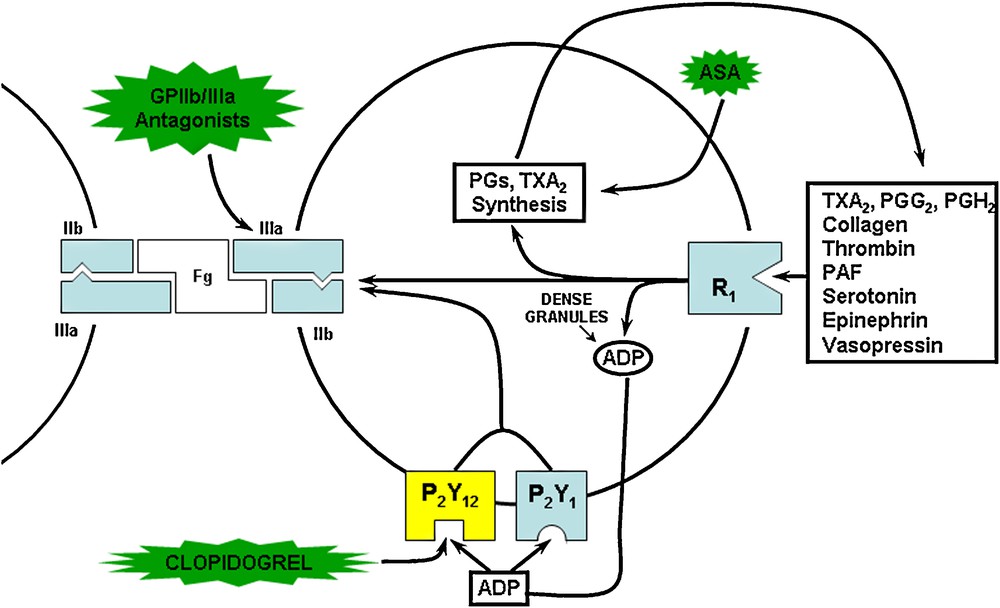
The active metabolite of clopidogrel irreversibly and selectively blocks the P2Y12 receptor of ADP on platelets [8], thus inhibiting the activation of the glycoprotein IIb/IIIa pathway and the aggregation induced by this agonist. It also inhibits the aggregation triggered by low concentrations of other mediators such as collagen and thrombin because it blocks the amplification of the aggregation by the ADP released from the dense granules. However, at high concentrations of these agonists, this amplification no longer plays an important role and the anti-aggregating effect of clopidogrel is counteracted.
However, despite the huge scientific and technical environment available, these two drugs could not be discovered and developed today because of the following characteristics of these drugs.
2 Ticlopidine and clopidogrel are prodrugs, which need an active metabolic transformation to affect their platelet target and develop their anti-aggregating activity
These two compounds were historically detected through a phenotypic screening (ADP-induced platelet aggregation) ex-vivo, after single or repeated administration to rats. Fortunately, that species, in contrast with others such as the guinea pig, produces enough active metabolite in the liver to observe an antiplatelet effect. Later [7], we were able to produce and purify the clopidogrel active metabolite by incubating the prodrug or an intermediate metabolite with rat or human hepatic microsomes but the process was tedious; we obtained only tiny quantities of this very unstable metabolite which must be used immediately in the platelet assays or chemically transformed into a stable adduct with acrylonitrile to be spectroscopically characterized. Probably because of this chemical instability, we were unable to synthesize this active metabolite. In other words, these molecules cannot exist on the shelves of chemical libraries and cannot be handled for an in vitro high throughput screening. On the other hand, phenotypic in vivo or ex-vivo screening was abandoned many years ago by pharmaceutical companies because of the following drawbacks:
- • ultra-low screening throughput;
- • large quantities of products needed for administration to animals;
- • costs and ethical issues linked to animal use;
- • in contrast with target-based screening, phenotypic screenings can recruit compounds with different mechanisms of action and it is not an easy task to find the primary target for each of them.
Altogether, I do not see how ticlopidine and clopidogrel could be discovered today from scratch in an industry, which only relies on “rational design” or high throughput in vitro technologies, using preferably target-based assays. New reversible antagonists of the P2Y12 ADP receptor (e.g., ticagrelor, cangrelor and elinogrel) were recently found, using such approaches [11].
3 The active metabolite of ticlopidine and clopidogrel is a reactive species, which irreversibly affects its target
Because platelets of treated subjects still showed inhibition after repeated washings, it was proposed early on that the active metabolite of ticlopidine and clopidogrel irreversibly affects its target. This was also supported by the observation that platelet inhibition is still detectable several days after stopping oral dosing with these drugs, and that recovery of normal platelet function occurs at a rate consistent with platelet turnover. These findings soon indicated that the inhibitory effect is permanent; i.e. the treated platelets are affected for the remainder of their lifespan. It was suspected that such an irreversible effect was due to the formation of a covalent bond between the active metabolite and its hypothetical receptor. This was confirmed many years later when it was found that the active metabolite contains a free thiol group which inactivates the P2Y12 ADP receptor through formation of a disulfide bridge with extracellular cysteine residues [12,13], most probably within the first extracellular loop of the receptor [13]. Thus, the duration of the antagonist effect is determined by the rate of receptor turnover and not by the clearance of the free drug or active metabolite. This is associated with a long duration of action, which allows a once-a-day administration.
Nowadays, many drug discovery teams are reluctant to develop such “covalent drugs”. One reason is that a permanent effect could also be a drawback if serious adverse reactions due to an exaggerated pharmacological effect occur (for example, bleeding with antiplatelet agents) and if there is no way (e.g. antidote) to promptly neutralize the drug action. More disturbing is the fact that most of these active metabolites are electrophilic and unselective species, which can covalently bind other cellular, and/or circulating macromolecules, such as DNA or proteins. Although not all toxicological effect are attributable to reactive metabolites, and not all covalent binding events lead to a deleterious biological consequence, a body of evidence suggests that inadequate production and/or detoxification of these reactive metabolites can cause (idiosyncratic) toxicity. In this respect, several publications [14,15] compile the structural features (toxicophores) within molecules that are thought to be responsible for their toxic properties after metabolic activation. For example, thiophene rings can undergo P450-mediated oxidation to unstable and reactive thiophene-S-oxide or thiophene epoxide metabolites [16]. Some thiophene-containing drugs (the diuretic tienilic acid and the NSAIDs tenoxicam and suprofen) were removed from the market for unacceptable side effects and it is suspected that a link could exist between the metabolic transformation of the thiophene ring into reactive species and the observed toxicity. With such an argument, some R&D teams may disqualify today unsubstituted thiophene-containing molecules as drug candidates. Thus, they would reject ticlopidine and the well-tolerated clopidogrel as drug candidates without experimentally investigating their toxicity! The same comment could apply to other “covalent drugs” such as aspirin, penicillin antibiotics, omeprazole…
4 Ticlopidine and, especially, clopidogrel are extensively metabolized
Ticlopidine is metabolized extensively by the liver, with only 22% of a radiolabelled dose representing unchanged ticlopidine at Cmax, in man. The drug and its metabolites are eliminated in the urine and faeces.
Clopidogrel is metabolized much more after a repeated 75 mg oral dose. Plasma concentration of the parent compound is very low and generally below the quantification limit beyond 2 hours after dosing. The main circulating metabolite is the carboxylic acid derivative (Fig. 3), which has no effect on platelet aggregation. Its glucuronide is also observed. Several other metabolites were isolated (Fig. 3) but only one, named H4, was identified as the active metabolite of clopidogrel. The metabolic cascade leading to H4 starts with the CYP450-mediated oxidation of the thiophene ring. The resulting unstable S-oxide rearranges into a thiolactone (SR 121683), which is ring-opened to afford the unstable thiol H4. The formation of this S-oxide as an intermediate species is supported by the isolation of its Diels-Alder dimerization product and glutathione adducts. Such a metabolic pathway was also described for other thiophene-containing compounds [18]. However, one cannot exclude the formation of an epoxide thiophene, as observed with other thiophene derivatives, which could also rearrange into the same thiolactone [16]. In actual fact, in vitro, CYP-mediated opening of the thiolactone ring leads to four thiol isomers: two cis diastereomers, H3 and H4, with a Z configuration of the exocyclic double bond and two trans-diastereomers, H1 and H2, with an E configuration of that double bond [7,17,18]. Since H1 and H2 were not measurable in plasma samples from clopidogrel-treated subjects [17,18] and the H3 metabolite was inactive [7a,17], it can be concluded that the H4 isomer is the clinically relevant clopidogrel active metabolite.
The identification of the enzymes involved in the “activating pathway” was also tricky. To cut a long story short, it was shown, mainly from in vitro experiments, that several CYPs (mainly CYP1A2, CYP2B6, and CYP2C19) [19] can mediate the first step, i.e. the formation of the thiolactone. More surprisingly, it was found that the second step, which could result from a simple hydrolytic ring opening, was also CYP-dependent. It was indeed shown recently [20] that incubations of the thiolactone with microsomes containing recombinant human P450 3A4, 3A5, 2C8, 2C9, 2C19, 2D6 or 1A2, lead to a sulfenic acid which after reaction with thiol nucleophiles present in the milieu (e.g. glutathione) produces mixed disulfides and, finally, H4 [20] (Fig. 5). The formation of the sulfenic acid intermediate was evidenced by trapping it with dimedone and characterizing the corresponding adduct by NMR and mass spectroscopy.
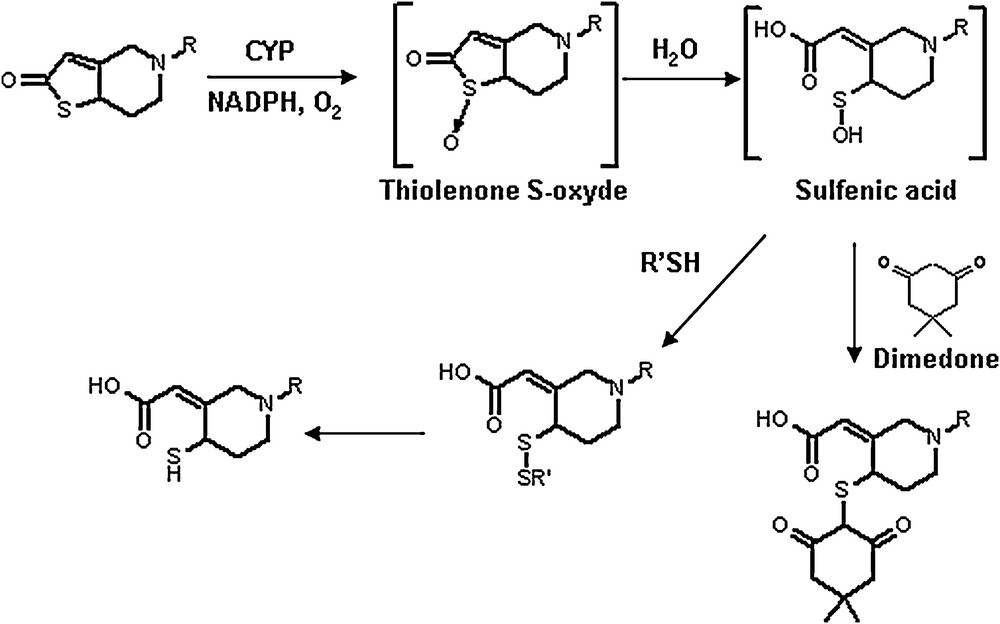
Involvement of a sulfenic acid intermediate in cytochrome P450 catalysed oxidation of ticlopidine and clopidogrel [20]. The existence of this unstable intermediate is demonstrated by the formation of a stable adduct with dimedone.
More recently, however, Bouman et al. [10] claimed that paraxonase-1 (PON-1), an esterase synthesized in the liver and associated with HDL in blood, is the rate-determining enzyme for the formation of H4 from the thiolactone. Moreover, the authors found that PON-1 QQ192 homozygous patients treated with clopidogrel after stent implantation, showed a “considerably higher risk than RR192 homozygous individuals of stent thrombosis, lower PON-1 plasma activity, lower plasma concentration of active metabolite and lower platelet inhibition”. Strangely enough, these pharmacokinetic, pharmacodynamic and clinical findings could not be confirmed in nine subsequent studies [18,21–28]. Moreover, it was recently reported [18,29] that PON-1 can indeed open the thiolactone ring but the resulting metabolite is an “endo” isomer (Fig. 3) in which the double bond has migrated from an exocyclic to an endocyclic position in the piperidine ring. As indicated [29], the HPLC system used by Bouman et al. [10] may not in fact have separated the active metabolite H4 and the “endo” isomer, thus leading to a wrong conclusion. The antiplatelet activity of this previously detected [17] minor “endo” metabolite has not been precisely evaluated yet, but it was reported that its AUC in healthy volunteers does not correlate with the antiplatelet response [18].
It is frequent that enzymes, operating in in vitro assays with microsomes or recombinant CYPs, play no role or just a limited one in vivo. Taking into account all the published clinical studies in man, it appears that CYP3A4/5 and CYP2C19 have the greatest role and are responsible for oxidation of clopidogrel to its active metabolite [19,30]. CYP2C19 could be the most important CYP for the conversion of clopidogrel to the intermediate thiolactone and CYP3A4 for the conversion of this thiolactone to the active metabolite H4. Not surprisingly, drug-drug interactions were found with co-administration of drugs, which inhibit or induce these CYPs [31]. Thus, the “active” metabolism of clopidogrel is inhibited by the CYP3A4 inhibitor, ketoconazole, and induced by rifampin. The lipophilic atorvastatin (a 3A4 substrate and inhibitor) was also reported to decrease the antiplatelet effect of clopidogrel in man. However, other studies were not able to confirm this result and there was no evidence that this potential pharmacodynamic interaction has clinical significance. Interestingly, hydrophilic statins do not affect the antiplatelet activity of clopidogrel and, thus, they could be more confidently prescribed in combination with it. Reduced antiplatelet effects of clopidogrel in patients co-administered with omeprazole, a proton pump inhibitor (PPI) which is a metabolism-dependent inhibitor of CYP2C19, emphasized the importance of this enzyme in the formation of the active metabolite [32]. However, no interaction or just a limited one occurred with two other PPIs, lanzoprazole and pantoprazole [33]. Whereas previous observational studies produced discordant results, a recent randomized trial [34] could not find an increased cardiovascular risk in patients receiving clopidogrel in combination with omeprazole. This did not prevent the FDA recently publishing a warning against the concomitant use of clopidogrel and omeprazole, emphasizing that this recommendation applies only to omeprazole and not to all PPIs; they also mentioned that pantoprazole may be an alternative PPI for consideration.
Clopidogrel was also found less antiaggregant in patients with a loss-of-function CYP2C19*2 polymorphism [31,35]. Subsequently, other clinical trials and a meta-analysis [36] reported that among patients treated with clopidogrel for PCI, carrying of even one reduced-function CYP2C19 allele appears to be associated with a significantly increased risk of major adverse cardiovascular events, particularly stent thrombosis. In March 2010, the FDA added a Boxed Warning to the label for clopidogrel to caution that this drug at recommended doses forms less active metabolite and has a lesser effect on platelet function in patients who are CYP2C19 poor metabolizers (it was estimated [36] that 2 % (homozygotes) to 26 % (heterozygotes) of the population are poor metabolizers). FDA also added that poor metabolizers with acute coronary syndrome or undergoing PCI treated with clopidogrel exhibit higher cardiovascular event rates than do patients with normal CYP2C19 function. Moreover, they informed that tests are available to identify patient's CYP2C19 genotype and can be used as an aid in determining therapeutic strategy. They also recommended considering alternative treatment or treatment strategies in patients identified as CYP2C19 poor metabolizers. However, the American Heart Association and American College of Cardiologists immediately argued that there was insufficient evidence for this warning. More recently, the first clinical analysis and the resulting FDA recommendations were also strongly challenged [37] by two other meta-analysis which concluded that although there was an association between the CYP2C19 phenotype and clopidogrel responsiveness, overall there was no significant association of phenotype with cardiovascular events (with the possible exception of stent thrombosis). Let us hope that ongoing trials will soon dissipate this dispute and provide a clear answer on the clinical utility of incorporating genetics and platelet function testing into treatment decisions.
Even today, most drug discovery teams would not be worried about the rapid and intense metabolization of clopidogrel. In fact, this is somehow compensated by the irreversible action of the active metabolite on platelets, which allows a once-a-day administration. It could even be argued that the rapid clearance of the unchanged drug and its active metabolite is an advantage because it minimizes off-target interactions, which could conceivably reduce non-mechanism-based toxicity. However, after simple experiments routinely performed today during the discovery phase, some R&D groups would be discouraged by the fact that the two main enzymes -CYP2C19 and CYP3A4-involved in the active transformation of the drug can be strongly affected by loss-of-function polymorphisms (CYP2C19) or co-administered drugs which induce or inhibit (competitively or irreversibly) them. Thus, the potential for drug-drug interaction and inter-individual response variability could be dissuasive to select such a product for development even if it appears, with use, that these drawbacks exist but are manageable.
5 Conclusion
Ticlopidine and clopidogrel cannot be discovered today through in vitro high throughput screenings because they must be transformed into an active metabolite, which is very unstable and cannot be stored on shelves. Moreover, its structure cannot be predicted, ab initio, by rational drug design. In other words, only an in vivo screening can detect these drugs. However, such screenings have not been used for decades in the pharmaceutical industry. And even if these molecules were discovered by chance, it is highly probable that they would be disqualified by the multiple computational models used today to predict ADMET properties. Alternatively, based on preliminary experimental results, some R&D teams would be very reluctant to develop such heavily metabolized drug candidates with reactive metabolites, which bind proteins. They would be also concerned by the fact that the active metabolite formation depends on enzymes, which can be either induced or inhibited by some co-administered drugs, thus leading to potentially deleterious drug-drug interactions. Moreover, one of these enzymes (CYP2C19) presents a loss-of-function polymorphism in a significant proportion of the population thus producing clopidogrel-resistant patients, at least in terms of antiplatelet activity.
Other “old” major drugs would not be discovered today for similar or different reasons (e.g., not fully elucidated multi-target mechanism of action). Conversely, important drugs recently discovered using modern technologies could not have been detected in the past. Thus, there is nothing to regret except that, up until now, efficiency for finding innovative and useful new drugs has not dramatically increased despite the recent genomic and technologic revolution!
Finally, at least two lessons could be learned from this ticlopidine and clopidogrel story. First, more attention should be paid to phenotypic screenings which now use whole cells, to complement the easier-to-perform target-based screenings. Secondly, one should not abandon drug candidates too quickly because of certain drawbacks encountered, which could finally be manageable. The prime consideration should be the benefit/risk ratio for the patients.