

1 Introduction
The coordination chemistry of 2,6-(Me2NCH2)2C5H3N (1, N’NN’) and related ligands was investigated mainly in relation to the potential application in catalysis of the corresponding metal complexes [1]. Several metal ions were used to investigate the coordination ability of 1, e.g. FeII [1f,1g], RuII [1a–d,g,2], RuIII [1d], RuIV [3], PdII [4], CuI [5b], CuII [5], UVI [6], ZnII [7]. By contrast, the ligand 2,6-(PhSeCH2)2C5H3N (4, SeNSe) (Scheme 1) was recently reported [8a] and only few of its PdII and RuII complexes were described as highly active catalysts for Heck coupling (C–C) reaction [8a] and oxidation of primary and secondary alcohols [8b], respectively. In most cases both ligands 1 and 4 were found to act as monometallic triconnective (“pincer”, Scheme 1, A) units, i.e. with both the sp2-hybridized nitrogen atom of the central pyridine ring and the donor atoms from two flexible pendant arms coordinated to the same metal atom. In only one case a different coordination pattern was described, i.e. the cation of the copper(I) complex [Cu(N’NN’)2][CuCl2] contains both ligand units chelating the metal atom through the pyridine nitrogen atom and the nitrogen of one pendant arm, while the other pendant arm is not coordinated to a metal centre (Scheme 1, B). This suggests that in addition to a pincer pattern such ligands might act as a bridge, the “free” pendant arm being involved in coordination to a second metal atom (Scheme 1, C) and thus providing the possibility for metal-metal interactions. For coinage metals such metalophilic interactions can influence the properties of the complexes, e.g. the optical properties [9].
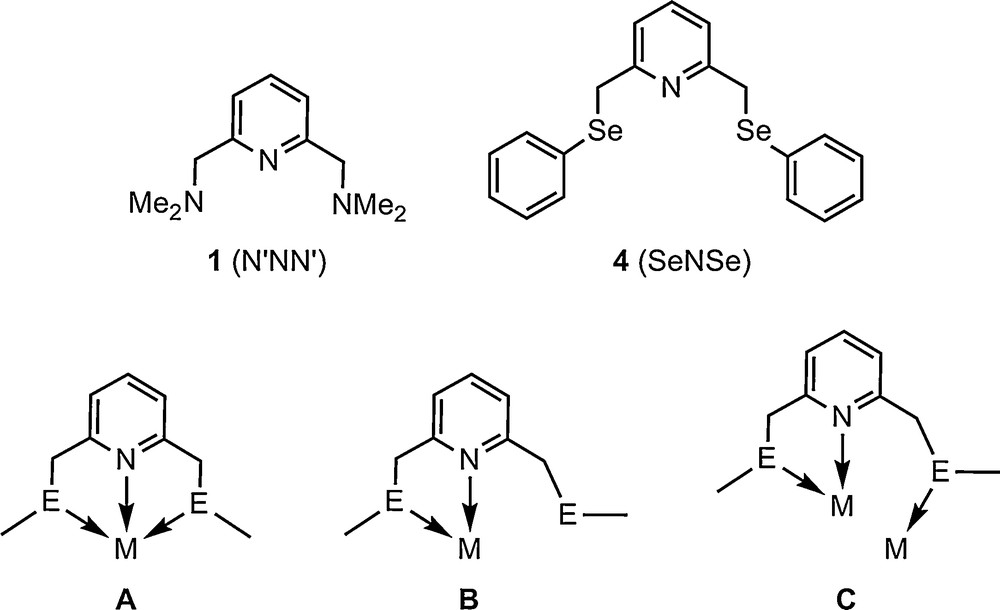
Ligands 1 and 4 and potential coordination patterns.
With respect to other nitrogen donor ligands related to 1, including macrocycles containing the fragment 2,6-substituted pyridine, in addition to metals as MnII [1g,10], FeII [1g,11], RuII, CoII [1g], CuII [5b,11,12], ZnII [11,13,14] or PbII [13b], several AgI complexes were investigated and their structure reported [10–13,15].
We describe herein the synthesis and spectroscopic characterization of some new coinage metal complexes containing the potential tridentate ligands N’NN’ (1) and SeNSe (4) as well as the structure of the tetranuclear silver(I) species [Ag4(N’NN’){O(O)CCF3}4(EtOH)] which shows intermetallic contacts and a bimetallic triconnective N’NN’ ligand.
2 Results and discussion
The potential tridentate ligand 2,6-(Me2NCH2)2C5H3N (1, N’NN’) was prepared according to a published method [4b], while for the synthesis of the related selenium-containing ligand 2,6-(PhSeCH2)2C5H3N (4, SeNSe) a slightly modified procedure [8a] was used. Reactions of 1 with [AgO(O)CCF3] (1:1 molar ratio) and [Ag(PPh3)(OTf)] (1:2 molar ratio) in anhydrous CH2Cl2 afforded the isolation of compounds [Ag(N’NN’){O(O)CCF3}] (2) and [{Ag(PPh3)}2(N’NN’)](OTf)2 (3) as light yellow or white-cream solids, respectively (Scheme 2). Both complexes were stable to air and moisture for long periods of time.
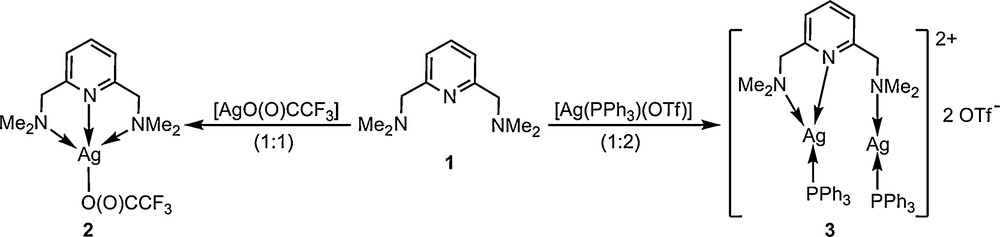
Synthesis of silver complexes 2 and 3.
The organoselenium(II) derivative 4 was reacted with H[AuCl4]·3H2O, [Ag(PPh3)(OTf)] or [Cu(MeCN)4]PF6 to yield 1:1 complexes [Au(SeNSe)Cl]Cl2 (5), [Ag(PPh3)(SeNSe)](OTf) (6) and [Cu(MeCN)(SeNSe)](PF6) (7) in good yields. When 4 was treated with [Cu(MeCN)4]PF6 in 2:1 molar ratio the complex [Cu(SeNSe)2](PF6) (8) was isolated (Scheme 3). All complexes containing 4 as a ligand were isolated as stable yellow (5, 7 and 8) or white solids (6). Analytical and spectroscopic data (IR and multinuclear NMR spectroscopies, mass spectrometry) were consistent with the anticipated formulas for the new complexes. The assignment of the 1H and 13C resonances in the room temperature NMR spectra (recorded in CDCl3 solutions) was based on the 2D experiments.
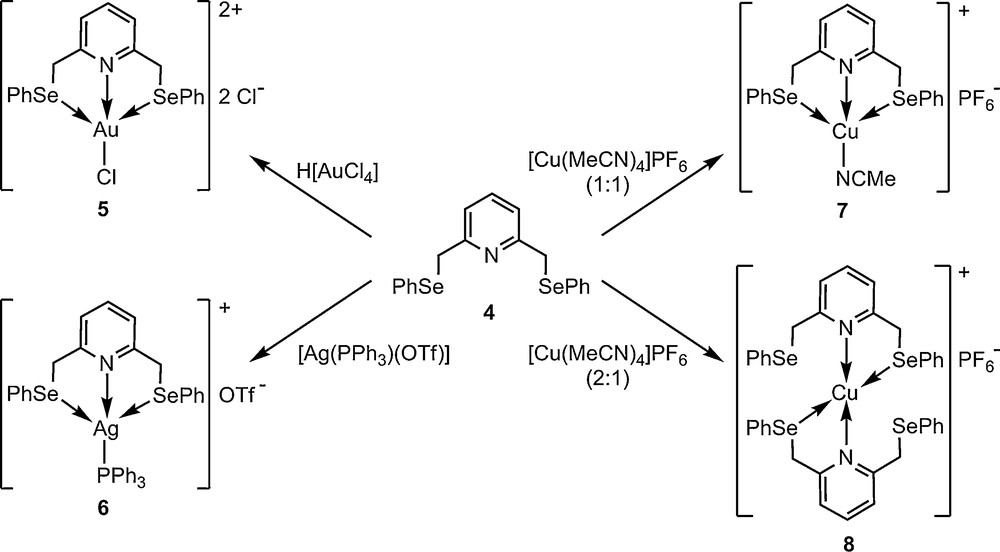
Synthesis of metal complexes 5–8.
The molecular ions for complexes 2, 3 and 5–8 were not observed in the MALDI(+) or ESI(+) mass spectra, thus suggesting easy splitting of the corresponding anionic group. The MALDI(+) spectra of 2 and 5 show the fragment [Ag(N’NN’)+] (m/z 301) and [Au(SeNSe)+] (m/z 616) as base peaks. For the silver complexes 3 and 6 the base peak corresponds to [Ag(PPh3)2+] (m/z 631), but for both complexes the spectra contain also the peaks assigned to [Ag(PPh3)(N’NN’)+] (m/z 562) and [Ag(PPh3)(SeNSe)+] (m/z 788), respectively. The ESI(+) mass spectrum of the copper complex 8 exhibit the metal-containing fragments [Cu(SeNSe)2+] (m/z 901) and [Cu(SeNSe)+] (m/z 482), the later one being the base peak for the complex 7 too.
The IR spectrum of 2 shows strong to medium bands at 1679, 1666 cm−l [νas(CO2)] and 1457 cm−l [νs(CO2)]. Although there is no clear difference between the Δ values [Δ = νas(CO2)–νs(CO2)] calculated for 3 (av. 215 cm−l) vs. the ionic NH4(O2CCF3) [1667 cm−l for νasym(CO2) and 1465 cm−l for νsym(CO2); Δ = 202 cm−l] [16], it is difficult to draw structural conclusions based only on the IR data. Apparently, the IR data seem to be consistent with a triflate anion covalently bound and ionic in nature in the silver complexes 3 and 6, respectively, in solid state [17]. However, in the absence of a single-crystal X-ray diffraction study it is difficult to clearly decide between these possibilities.
The 1H and 13C NMR spectra for complexes 2 and 3 are very similar, with respect to the aliphatic and aromatic resonances, to those of the free ligand 1. The sharp singlet resonances observed for the protons and carbon atoms of the methylene and methyl groups suggest the equivalence of the pendant arms of the N’NN’ ligand unit, at room temperature, on the NMR time scale. For complex 3 this suggests a dynamic behavior, the silver atoms of a cation unit being alternatively coordinated by two nitrogen atoms. This dynamic process is also supported by the presence of only one set of broader 13C resonances for the phenyl groups of the PPh3 ligands (phosphorus-carbon coupling observed, see Experimental Section). In addition to the expected 13C resonances for the neutral ligands, the spectra of both compounds 2 and 3 contain resonances for the anions, with a quartet pattern due to carbon-fluorine coupling, i.e. δ 117.53 [1J(CF) 291.3 Hz, CF3] and 162.36 [2J(CF) 34.8 Hz,–C(O)O–] for 2, and δ 120.57 [q, 1J(CF) 320.8 Hz, CF3] for 3. The 19F NMR spectrum of 3 exhibits only one singlet resonance (δ–77.9 ppm), consistent with the presence of only one species in solution.
Similar considerations can be drawn out from the 1H and 13C NMR spectra for metal complexes of the selenium-containing ligand 4, although the 77Se resonance could be observed only for the free ligand. Only one set of 1H and 13C resonances was observed for the two pendant arms (e.g. singlet resonances for all methylene protons or carbons). Also the protons in positions 3 and 5 of the pyridine ring gave one doublet resonance in the aromatic region. This suggests either a symmetrical (Se,N,Se-monometallic triconnective) coordination in the case of complexes 5–7 or a fast dynamic process, at room temperature, for compound 8, with the copper atom being coordinated by the nitrogen of the pyridine ring and alternating selenium atom from pendant arms [Cu(NSe)2 core]. For complexes 6 and 7 the 1H and 13C NMR spectra exhibit, in addition to the resonances for ligand 4, further signals for the PPh3 and MeCN molecules coordinated to the metal atom. The presence of the OTf− and PF6− anions was proved by 19F and 31P NMR spectra; the presence of only one resonance with expected multiplicity pattern being consistent with the presence of only one species in solution.
In case of complexes 3 and 6, which both contain PPh3 coordinated to the metal atom, the 31P NMR spectroscopy was a very useful tool for the investigation of the coordination environment of the silver. For compound 3, the 31P NMR spectrum, recorded at room temperature, exhibits two broad resonances at δ 11.9 and 16.0 ppm. This might be due to a dissociative process involving cleavage of N-Ag(PPh3), as mentioned above. Such dissociative processes are well-documented in the case of phosphine complexes of coinage metals [18]. At –55 °C the 31P NMR spectrum of 3 shows two resolved resonances, each of them with a pattern of doublet of doublets due to the couplings with 107Ag and 109Ag isotopes, thus indicating the presence of two different environments for silver-phosphine cores. The coordination number (CN) around a silver atom in a silver-phosphine species can be estimated on the basis of the magnitude of the silver-phosphorus coupling constant. As mentioned in literature, the 1J(107Ag,P) coupling constants are inversely proportional to the coordination number of the silver atom [19] and increase with decreasing of Ag-P distances [20]. Thus, using the magnitude of the observed coupling constants the resonance at δ 11.0 [1J(P,107Ag) 511.3 Hz] was assigned to a Ag(PPh3) fragment coordinated by two nitrogen atoms [N2Ag(PPh3) core, CN = 3)], while the resonance at δ 14.4 [1J(P,107Ag) 627.6 Hz] was assigned to a Ag(PPh3) fragment coordinated by one nitrogen atom [NAg(PPh3) core, CN = 2], respectively. On the basis of similar considerations, it can be assumed that at –55 °C the 31P NMR spectrum of 6 shows resonances, with doublet of doublet pattern, which can be assigned to two different silver-containing species in solution. The resonance at δ 7.9 [1J(P,107Ag) = 398.0 Hz] can be assigned to a tetracoordinated species in which the Ag(PPh3) fragment is coordinated by all donor atoms of a SeNSe ligand, while the less resolved resonance at δ 11.5 [1J(P,107Ag) = 551.6 Hz] might be due to a tricoordinated species in which a silver-selenium bond was dissociated.
The complex 3 exhibits an intense, long-lived luminescence in the solid state with emission energies in the green region of the visible spectrum. It is emissive in the solid state both at room temperature and in frozen solution (an intermediate situation between solid and solution state), as shown in Fig. 1. Emissions in the solid state appear near 488 nm, while excitation near 425 nm. These maxima appear shifted to higher energies in dichloromethane frozen solutions, i.e. 525 nm for emission and 475 nm for excitation, respectively. In all cases only one band is observed.
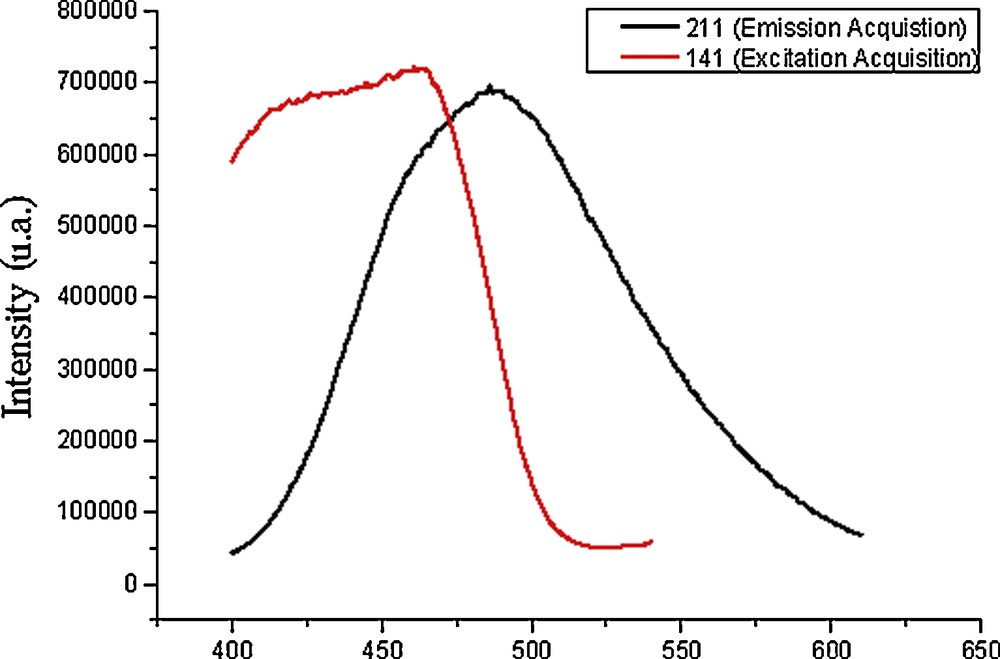
Emission and excitation spectra for compound 3 in solid state at room temperature.
In an attempt to grow single crystals suitable for X-ray diffraction studies compound 2 was dissolved in CH2Cl2 and hexane was slowly added (1:5, v/v). The X-ray diffraction analysis of one of the few colorless crystals obtained in this way revealed the molecular structure of an unexpected polynuclear silver species, [Ag4(N’NN’){O(O)CCF3}4(EtOH)]n (2a). The presence of the ethanol molecule can be explained by presence of this solvent in the methylene chloride used to grow the crystals. No attempts to prepare this polymer in a higher yield were carried out so far. Selected interatomic distances and angles are listed in Table 1.
Selected bond distances [Å] and angles [°] for compound 2a.a
| Ag(1)–O(1) | 2.235(7) | Ag(3)–O(4) | 2.16(2) |
| Ag(1)–N(1) | 2.334(7) | Ag(3)–O(6) | 2.693(6) |
| Ag(1)–N(2) | 2.416(7) | Ag(3)–O(6a’)a | 2.595(6) |
| Ag(1)–Ag(2) | 3.075(5) | Ag(3)–N(3) | 2.300(7) |
| Ag(2)–O(2) | 2.346(7) | Ag(4)–O(2) | 2.684(8) |
| Ag(2)–O(3) | 2.23(2) | Ag(4)–O(5) | 2.496(7) |
| Ag(2)–O(5) | 2.473(7) | Ag(4)–O(7) | 2.234(7) |
| Ag(2)–O(9) | 2.485(10) | Ag(4)–O(8) | 2.240(8) |
| Ag(2)–Ag(3) | 3.009(4) | Ag(4)–Ag(4a)a | 2.889(4) |
| C(13)–O(1) | 1.279(11) | C(17)–O(5) | 1.254(9) |
| C(13)–O(2) | 1.264(10) | C(17)–O(6) | 1.243(8) |
| C(15)–O(3) | 1.22(2) | C(19)–O(7) | 1.212(10) |
| C(15)–O(4) | 1.32(2) | C(19)–O(8a)a | 1.252(10) |
| C(21)–O(9) | 1.371(15) | ||
| N(1)–Ag(1)–N(2) | 76.3(3) | N(3)–Ag(3)–O(6a’) | 101.0(3) |
| O(1)–Ag(1)–N(1) | 129.3(2) | O(6a’)–Ag(3)–O(4) | 78.8(6) |
| O(1)–Ag(1)–N(2) | 129.0(3) | O(4)–Ag(3)–Ag(2) | 75.4(6) |
| Ag(2)–Ag(1)–N(1) | 116.8(2) | Ag(2)–Ag(3)–N(3) | 107.70(19) |
| Ag(2)–Ag(1)–N(2) | 130.6(2) | N(3)–Ag(3)–O(4) | 173.2(7) |
| Ag(2)–Ag(1)–O(1) | 81.5(2) | O(6a’)–Ag(3)–Ag(2) | 143.27(14) |
| O(6)–Ag(3)–N(3) | 99.3(2) | ||
| O(6)–Ag(3)–O(4) | 87.5(7) | ||
| O(6)–Ag(3)–O(6a’) | 78.4(2) | ||
| O(6)–Ag(3)–Ag(2) | 74.8(1) | ||
| O(9)–Ag(2)–Ag(3) | 173.7(2) | O(7)–Ag(4)–O(8) | 162.8(2) |
| O(9)–Ag(2)–O(2) | 82.2(3) | O(7)–Ag(4)–O(2) | 95.9(3) |
| O(9)–Ag(2)–O(3) | 95.7(7) | O(7)–Ag(4)–O(5) | 98.7(2) |
| O(9)–Ag(2)–O(5) | 100.5(3) | O(7)–Ag(4)–Ag(4a) | 83.34(18) |
| Ag(3)–Ag(2)–O(2) | 100.08(19) | O(8)–Ag(4)–O(2) | 95,4(2) |
| Ag(3)–Ag(2)–O(3) | 79.2(7) | O(8)–Ag(4)–O(5) | 96.6(2) |
| Ag(3)–Ag(2)–O(5) | 85.61(19) | O(8)–Ag(4)–Ag(4a) | 79.51(18) |
| O(2)–Ag(2)–O(3) | 146.1(7) | O(2)–Ag(4)–O(5) | 76.9(2) |
| O(2)–Ag(2)–O(5) | 83.9(2) | O(2)–Ag(4)–Ag(4a) | 125.03(15) |
| O(3)–Ag(2)–O(5) | 129.3(6) | O(5)–Ag(4)–Ag(4a) | 157.88(14) |
| O(2)–Ag(2)–Ag(1) | 73.69(18) | ||
| O(3)–Ag(2)–Ag(1) | 74.2(7) | ||
| Ag(3)–Ag(2)–Ag(1) | 70.63(13) | ||
| O(5)–Ag(2)–Ag(1) | 143.35(14) | ||
| O(9)–Ag(2)–Ag(1) | 104.7(3) | ||
| C(2)–N(1)–Ag(1) | 111.8(5) | C(6)–N(1)–Ag(1) | 125.6(5) |
| C(7)–N(2)–Ag(1) | 102.0(5) | C(10)–N(3)–Ag(3) | 113.0(4) |
| C(8)–N(2)–Ag(1) | 115.2(6) | C(11)–N(3)–Ag(3) | 108.9(5) |
| C(9)–N(2)–Ag(1) | 111.1(6) | C(12)–N(3)–Ag(3) | 108.3(5) |
| C(13)–O(1)–Ag(1) | 117.4(6) | C(17)–O(5)–Ag(2) | 121.1(5) |
| C(13)–O(2)–Ag(2) | 120.9(6) | C(17)–O(5)–Ag(4) | 131.9(5) |
| C(17)–O(6)–Ag(3) | 123.5(6) | ||
| C(17)–O(6)–Ag(3a’) | 134.8(5) | ||
| C(15)–O(3)–Ag(2) | 120.3(15) | C(19)–O(7)–Ag(4) | 122.2(6) |
| C(15)–O(4)–Ag(3) | 128.0(13) | C(19)–O(8a)–Ag(4) | 125.9(6) |
| C(21)–O(9)–Ag(2) | 132.3(11) | ||
| Ag(2)–O(2)–Ag(4) | 93.8(2) | Ag(3)–O(6)–Ag(3a’) | 101.6(2) |
| Ag(2)–O(5)–Ag(4) | 95.6(2) | ||
| O(1)–C(13)–O(2) | 130.5(8) | O(5)–C(17)–O(6) | 127.2(8) |
| O(3)–C(15)–O(4) | 123.0(16) | O(7)–C(19)–O(8a) | 128.9(9) |
a Symmetry equivalent atoms (1 − x, 2 − y, −z) and (2 − x, 1 − y, −z) are given by “prime” and “a”, respectively.
The crystal of 2a contains a polymer arrangement built from repeating tetranuclear units of the type depicted in Fig. 2. In such a tetranuclear unit the N’NN’ ligand act as a bimetallic triconnective unit, bridging Ag1 [Ag(1)–N(1) 2.334(7) Å, Ag(1)–N(2) 2.416(7) Å] and Ag3 atoms [Ag(3)–N(3) 2.300(7) Å]. The Ag–N bond distances are slightly different, nevertheless the average of these bond distances compare well with those found in the literature [10–13,15]. The distance between the silver atoms bridged by the N’NN’ ligand is too long [Ag(1)····Ag(3) 3.51(4) Å; cf. sums of the corresponding van der Waals radii, ΣrvdW(Ag,Ag) 3.4 Å [21]] to suggest any metal-metal interaction. However, silver-silver contacts are established with a third metal atom, Ag2 [Ag(1)–Ag(2) 3.075(5) Å, Ag(2)–Ag(3) 3.009(4) Å]. The metal-metal interactions in the resulting angular Ag3 system are supported by three bridging trifluoroacetate units: two of them between Ag2 and Ag3 atoms, while the third one bridge Ag2 and Ag1 atom (coordinated by two nitrogen atoms of the N’NN’ ligand). The oxygen atoms O2 and O5, from two trifluoroacetate ligands bridging different silver atoms of the Ag3 system, are involved in coordination to the forth metal atom of the tetranuclear fragment [Ag(4)–O(2) 2.684(8) Å, Ag(4)–O(5) 2.496(7) Å]. As a result these trifluoroacetate ligands exhibit each a trimetallic triconnective pattern. The Ag2O2 four-membered ring thus formed is not planar, but folded [dihedral angle O(2)Ag(2)O(5)/O(2)Ag(4)O(5) 31.5°]. However this folding is not enough to bring in contact Ag2 and Ag4 atoms [Ag(2)····Ag(4) 3.679(5) Å].
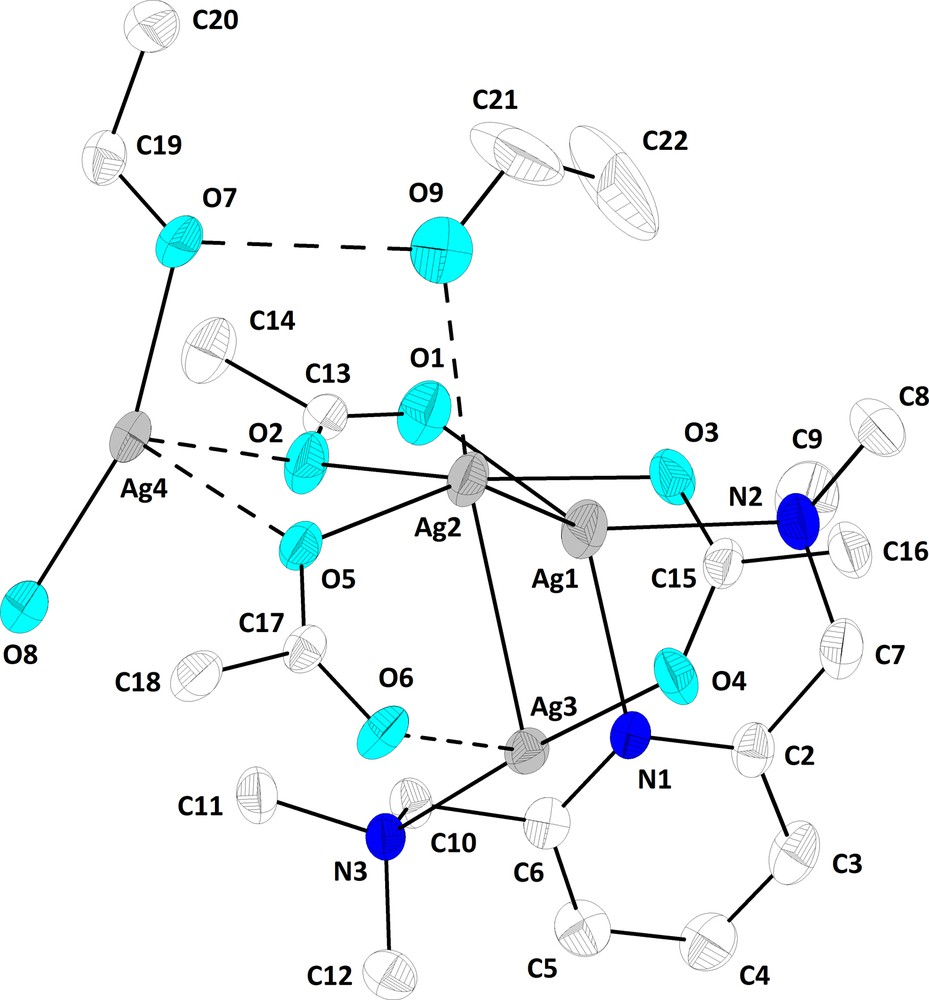
ORTEP representation at 20% probability and atom numbering scheme for the tetranuclear unit in 2a (hydrogen and fluorine atoms are omitted for clarity).
This Ag4 atom is part of the central [Ag2{O(O)CCF3}2] system of the centrosymmetric dimer built from two tetranuclear fragments (Fig. 3). The transanular Ag(4)–Ag(4a) of 2.889(4) Å, established between the metal atoms of the central Ag2O4C2 eight-membered ring, suggests a strong silver-silver interaction. This distance is considerably shorter than observed in the similar Ag2O4C2 eight-membered rings of the polymer [Ag2{O(O)CCF3}2]n [Ag–Ag 2.973(1) Å] [22].
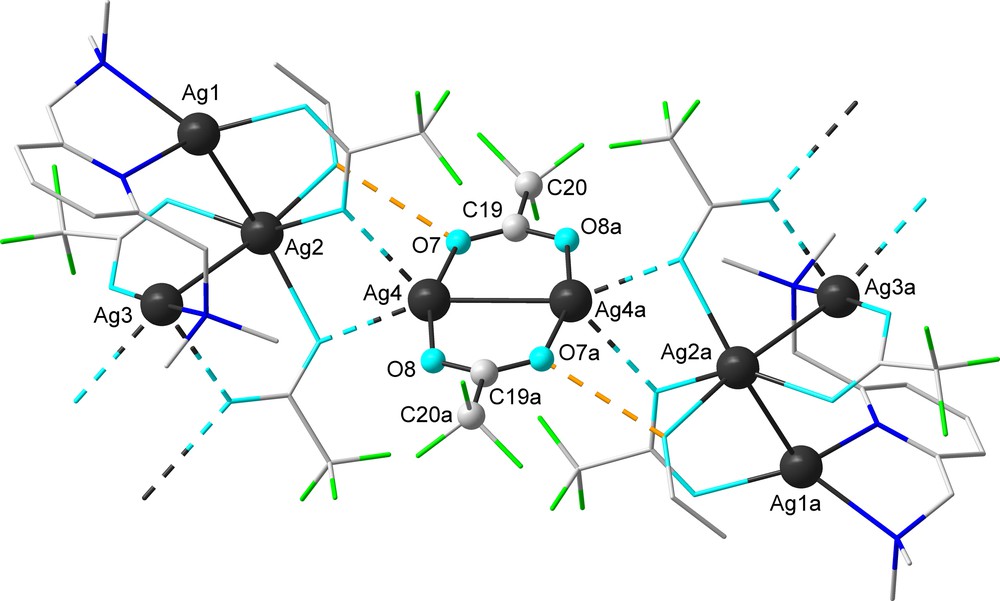
View of a centrosymmetric dimer built from tetranuclear units through bridging trifluoroacetate anions in the crystal of 2a (hydrogen atoms are omitted for clarity) [symmetry equivalent atoms (2–x, 1–y,–z) are given by “a”].
An ethanol molecule is coordinated through its oxygen to the Ag2 atom, the vector of this bond being placed in trans to the Ag3 atom [O(9)–Ag(2)–Ag(3) 173.7(2)°]. A hydrogen bond between the hydroxyl group of the alcohol molecule and the O7 atom is suggested by the distance O(9)····O(7) 2.93 Å.
The dimer units are doubly connected into a polymer through Ag(3)–O(6a’) [2.595(6) Å] interactions, the resulting Ag2O2 four-membered ring being in this case planar (Fig. 4).
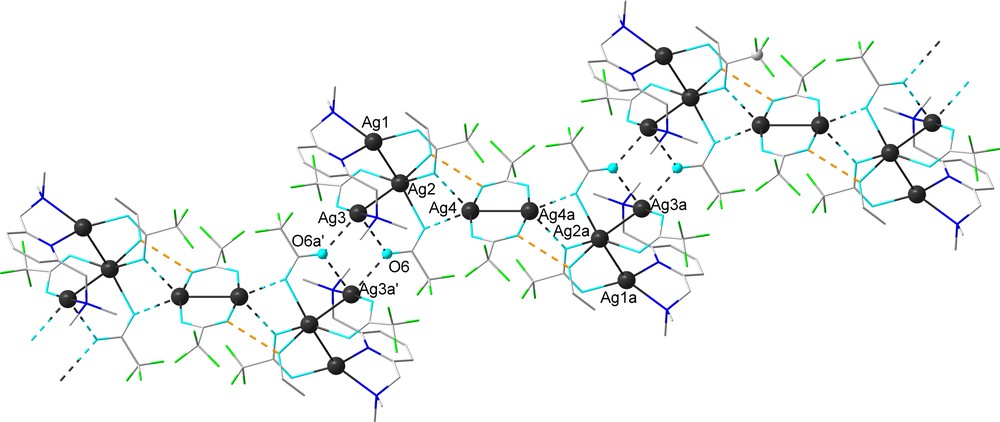
View of the polymer arrangement built from dimer units through Ag···O interactions in the crystal of 2a (hydrogen atoms are omitted for clarity) [symmetry equivalent atoms (2–x, 1–y,–z) and (1–x, 2–y,–z) are given by “a” and “prime”, respectively].
Taking into account all silver-nitrogen [range 2.300(7)–2.416(7) Å], silver-oxygen [range 2.16(2)–2.693(6) Å] and silver-silver [range 2.889(4)–3.075(5) Å] bonds and interactions per metal atom, four different overall coordination geometry around the silver atoms can be considered:
- • distorted tetrahedral Ag[N2OAg] core for Ag1;
- • distorted capped-trigonal bipyramidal Ag[O4Ag2] core for Ag2, with Ag1 capping the face described by Ag3, O2 and O3 atoms;
- • distorted square pyramidal Ag[NO3Ag] core for Ag3, with O6 in apical position;
- • distorted trigonal bipyramidal Ag[O4Ag] core for Ag4, with O7 and O8 atoms in axial positions [O(7)–Ag(4)–O(8) 162.8(2) Å].
3 Conclusions
As expected for most of the new coinage metal complexes reported here 2,6-(Me2NCH2)2C5H3N (N’NN’) (1) and 2,6-(PhSeCH2)2C5H3N (SeNSe) (4) act as pincer (monometallic triconnective) ligands. However, when an appropriate metal-to-ligand ratio was used a bridging (e.g. bimetallic triconnective) pattern can be achieved as in complex [{Ag(PPh3)}2(N’NN’)](OTf)2 (3). This compound was found to exhibit an intense luminescence in the solid state which might be due to potential metal-metal interaction. On the other hand in the copper(I) complex [Cu(SeNSe)2](PF6) (8) the ligand appears to have a free donor atom still available for coordination to a metal centre. In this case a reaction of 8 with an appropriate coordinatively unsaturated metal-containing moiety can provide new homo- or heterometallic polynuclear species. The ability of such ligands to act as bridging units between metal atoms was also supported by the unexpected polynuclear species, [Ag4(N’NN’){O(O)CCF3}4(EtOH)]n (2a), which contains metal-metal contacts and four different coordination environments around silver atoms.
4 Experimental
4.1 General considerations
All manipulations of air- and moisture-sensitive compounds were carried out in argon atmosphere using standard Schlenk techniques. Solvents were dried using standard procedures and freshly distilled under argon prior to use. Chemicals of commercial grade (Aldrich or Merck) were used as received. The ligand 2,6-(Me2NCH2)2C5H3N (1, N’NN’) was prepared according to a published procedure [4b]. The 1H, 13C, 19F and 31P NMR spectra were recorded at room temperature, in CDCl3, on Bruker Avance 300 (for 4) or Avance 400 spectrometers. The 1H and 13C chemical shifts are reported in ppm relative to the residual peak of the solvent as internal standard [ref. CHCl3: δ(1H) 7.26, δ(13C) 77.0 ppm]. The resonance position in the 19F and 31P NMR spectra were referenced to external CCl3F and H3PO4 (85%), respectively. The 77Se spectra were obtained using diphenyl diselenide as external standard. Chemical shifts are reported relative to dimethyl selenide (δ 0 ppm) by assuming that the resonance of the standard is at δ 461 ppm [23]. The NMR spectra were processed using the MestReNova software [24]. The ESI(+) mass spectra were recorded using an Esquire 3000 ion-trap mass spectrometer (Bruker Daltonic GmbH) equipped with a standard ESI/APCI source. The MALDI(+) mass spectra were recorded from CHCl3 solutions on a MALDI-TOF Microflex (Bruker) instrument (DCTB as matrix). Mass spectra were processed using the MASPEC II software [25]. Infrared spectra (4000–250 cm−1) were recorded with a Perkin-Elmer Spectrum One IR instrument in attenuated total reflection method (ATR).
4.2 Syntheses of compounds
4.2.1 [Ag{2,6-(Me2NCH2)2C5H3N}{O(O)CCF3}] (2)
Silver trifluoroacetate, [AgO(O)CCF3] (0.055 g, 0.25 mmol), was added to a solution of 1 (0.048 g, 0.25 mmol) in anhydrous CH2Cl2 (20 mL). The reaction mixture was stirred for 1 h at room temperature. The removal of the solvent in vacuum gave 2 as a light yellow solid, which was washed with hexane. Yield: 0.167 g (91%). 1H NMR (400 MHz, δ, ppm): 2.38 (s, 12H, NMe), 3.57 (s, 4H, CH2), 7.18 [d, 3J(HH) = 7.7 Hz, 2H, H3,5], 7.74 [t, 3J(HH) 7.7 Hz, 1H, H4]. 13C NMR (100.6 MHz, δ, ppm): 46.73 (s, NMe), 64.99 (s, CH2), 117.53 [q, 1J(CF) = 291.3 Hz, CF3], 123.46 (s, C3,5), 138.71 (s, C4), 156.96 (s, C2,6), 162.36 [q, 2J(CF) = 34.8 Hz,–C(O)O–]. MS [MALDI(+), m/z, %]: 300.1 (100) [Ag(N’NN’)+] [N’NN’ = 2,6-(Me2NCH2)2C5H3N]. IR (ATR, cm−1): 1679 (s), 1666 (s) [νas(CO2)], 1457 (m) [νs(CO2)], 423 (w), 404 (m) [ν(AgN)].
4.2.2 [{Ag(PPh3)}2{2,6-(Me2NCH2)2C5H3N}](OTf)2 (3)
Silver(triphenylphosphine) triflate [Ag(PPh3)(OTf)] (0.531 g, 1.02 mmol) was added to a solution of 1 (0.099 g, 0.512 mmol) in anhydrous CH2Cl2 (20 mL). The reaction mixture was stirred for 1 h at room temperature. After removal of the solvent in vacuum, the solid was washed with hexane to give 3 as a white-cream compound. Yield: 0.51 g (92%). 1H NMR (400 MHz, δ, ppm): 2.17 (s, 12H, NMe), 3.70 (s, 4H, CH2), 7.32 [d, 3J(HH) = 7.7 Hz, 2H, H3,5], 7.44 (m, 30H, PPh), 7.81 [t, 3J(HH) 7.7 Hz, 1H, H4]. 13C NMR (100.6 MHz, δ, ppm): 46.38 (s, NMe), 65.41 (s, CH2), 120.57 [q, 1J(CF) = 320.8 Hz, CF3], 123.61 (s, C3,5), 129.10 [d, 2J(CP) = 6.4 Hz, PPh-ortho], 130.56 [d, 1J(CP) = 34.3 Hz, PPh-ipso], 130.88 (s, PPh-para), 133.57 [d, 3J(CP) = 14.3 Hz, PPh-meta], 139.58 (s, C4), 157.17 (s, C2,6). 19F NMR (376.5 MHz, δ, ppm):–77.9 (s, 6F, CF3). 31P NMR (162 MHz, δ, ppm, r.t.): 11.9 (s,br), 16.0 (s,br). 31P NMR (121 MHz, δ, ppm,–55 °C): 11.0 [dd, 1J(P,107Ag) = 511.3, 1J(P,109Ag) = 562.8 Hz], 14.4 [dd, 1J(P,107Ag) = 627.6, 1J(P,109Ag) = 724.1 Hz]. MS [MALDI(+), m/z, %]: 631.3 (100) [Ag(PPh3)2+], 562.3 (53) [Ag(PPh3)(N’NN’)+] [N’NN’ = 2,6-(Me2NCH2)2C5H3N]. IR (ATR, cm−1): 1600 (vw), 1583 (vw) [ν(C = C)], 1479 (w), 1434 (m) [ν(C = N)], 1265 (s), 1242 (m) [νas(SO3)], 1221 (m) [νs(CF3)], 1150 (m) [νas(CF3)], 1024 (s) [νs(SO3)], 432 (w), 400 (w) [ν(AgN)].
4.2.3 2,6-(PhSeCH2)2C5H3N (4)
Compound 4 was obtained from Ph2Se2 (1 g, 3.2 mmol), NaBH4 (0.242 g, 6.4 mmol) and 2,6-bis(bromomethyl)pyridine (0.42 g, 3.2 mmol) in ethanol (40 mL) using a slightly modified procedure [8a]. The solvent was completely removed in vacuum from the reaction mixture and the remaining white solid was washed with hexane. Recrystallization from ethanol gave the title compound 4 as white crystals. Yield: 1.176 g (85%). 1H NMR (300 MHz, δ, ppm): 4.19 [s, 2J(HSe) = 12.4 Hz, 4H, CH2], 6.92 [d, 3J(HH) = 7.7 Hz, 2H, H3,5], 7.23 (m, 6H, SePh-meta + para), 7.38 [t, 3J(HH) 7.7 Hz, 1H, H4], 7.23 (m, 4H, SePh-ortho). 13C NMR (100.6 MHz, δ, ppm): 33.67 [s, 1J(CSe) = 62.5 Hz, CH2], 120.98 (s, C3,5), 127.27 (s, SePh-para), 128.96 (s, SePh-meta), 130.01 (s, SePh-ipso), 133.49 (s, SePh-ortho), 136.74 (s, C4), 158.36 (s, C2,6). 77Se NMR (100 MHz, δ, ppm): 368.3 (s). MS [MALDI(+), m/z, %]: 419.9 (20) [M+ + H]. IR (ATR, cm−1): 1576 (m), 1568 (m) [ν(C = C)], 1475 (m), 1436 (m) [ν(C = N)].
4.2.4 [Au{2,6-(PhSeCH2)2C5H3N}Cl]Cl2 (5)
HAuCl4·3H2O (0.011 g, 0.028 mmol) was added to a solution of 4 (0.012 g, 0.028 mmol) in anhydrous diethyl ether (20 mL). The reaction mixture was stirred for 30 minutes at room temperature, followed by removal of the solvent to dryness. The residue was solved in dry THF (20 mL), treated with NaHCO3 (0.047 g, 0.056 mmol) and then stirred for 24 h at room temperature. The solution was concentrated to a volume of 3 mL and hexane (5 mL) was added. A yellow oil separated and it was treated with Et2O and EtOH to give the title compound 5 as a light yellow solid. Yield: 0.020 g (96%). 1H NMR (400 MHz, δ, ppm): 4.64 [s, 4H, CH2], 6.92 [d, 3J(HH) = 7.7 Hz, 2H, H3,5], 7.36 [t, 3J(HH) = 7.4 Hz, 4H, SePh-meta], 7.44 [m, 3H, H4 + SePh-para], 7.66 [d, 3J(HH) = 7.6 Hz, 4H, SePh-ortho]. MS [MALDI(+), m/z, %]: 616.0 (100) [Au(SeNSe)+], 419.9 (67) [SeNSe+ + H] [SeNSe = 2,6-(PhSeCH2)2C5H3N]. IR (ATR, cm−1): 463 (m) [ν(AuN)], 330 (m) [ν(AuCl)].
4.2.5 [Ag(PPh3){2,6-(PhSeCH2)2C5H3N}](OTf) (6)
[Ag(PPh3)(OTf)] (0.034 g, 0.06 mmol) was added to a solution of 4 (0.028 g, 0.06 mmol) in anhydrous CH2Cl2 (15 mL). The reaction mixture was stirred for 1 h at room temperature. After removal of the solvent in vacuum, the solid was washed with hexane to give 6 as a white solid. Yield: 0.051 g (82%). 1H NMR (400 MHz, δ, ppm): 4.20 [s, 2J(HSe) = 13.8 Hz, 4H, CH2], 7.05 [d, 3J(HH) = 7.8 Hz, 2H, H3,5], 7.10 (m, 8H, SePh-ortho + meta), 7.20 (m, 2H, SePh-para), 7.30 (m, 12H, PPh-ortho + meta), 7.42 (m, 3H, PPh-para), 7.52 [t, 3J(HH) 7.7 Hz, 1H, H4]. 13C NMR (100.6 MHz, δ, ppm): 35.24 (s, CH2), 123.28 (s, C3,5), 127.55 (s, SePh-ipso), 128.02 (s, SePh-para), 129.16 [d, 2J(CP) = 9.9 Hz, PPh-ortho], 129.37 (s, SePh-meta), 130.79 (s, PPh-para), 130.95 [d, 1J(CP) = 32.4 Hz, PPh-ipso], 132.68 (s, SePh-ortho), 133.63 [d, 3J(CP) = 15.8 Hz, PPh-meta], 138.56 (s, C4), 157.01 (s, C2,6). 19F NMR (376.5 MHz, δ, ppm): −77.9 (s, 3F, CF3). 31P NMR (121.5 MHz, δ, ppm, −55 °C): 7.9 [dd, 1J(P,107Ag) = 398.0, 1J(P,109Ag) = 446.6 Hz], 11.5 [dd,br, 1J(P,107Ag) = 551.6, 1J(P,109Ag) = 597.0 Hz]. MS [ESI(+), m/z, %]: 787.7 [Ag(PPh3)(SeNSe)+], 631.0 [Ag(PPh3)2+ as base peak] [SeNSe = 2,6-(PhSeCH2)2C5H3N]. IR (ATR, cm−1): 1592 (w), 1568 (m) [ν(C = C)], 1478 (m), 1436 (m) [ν(C = N)], 1268 (m) [νas(SO3)], 1147 (m) [νs(CF3)], 1093 (m) [νas(CF3)], 1022 (m) [νs(SO3)], 395 (m) [ν(AgN)].
4.2.6 [Cu(MeCN){2,6-(PhSeCH2)2C5H3N}](PF6) (7)
To a solution of 4 (0.082 g, 0.2 mmol) in anhydrous CH2Cl2 (40 mL) [Cu(MeCN)4]PF6 (0.072 g, 0.2 mmol) was added in one portion. The reaction mixture was stirred at room temperature, under argon, for 2 h. Evaporation of the solvent in vacuum resulted in a yellow solid which was washed with hexane to gave the title compound 7. Yield: 0.106 g (87%). 1H NMR (400 MHz, δ, ppm): 2.22 (s, 3H, Meacetonitrile), 4.42 (s,br, 4H, CH2), 7.20 (m, 12H, H3,5 + SePh), 7.61 [t, 3J(HH) 7.3 Hz, 1H, H4]. 13C NMR (100.6 MHz, δ, ppm): 1.98 (s, Meacetonitrile), 38.44 (s, CH2), 117.12 (s, CNacetonitrile), 123.46 (s, C3,5), 127.01 (s, SePh-ipso), 128.73 (s, SePh-para), 129.68 (s, SePh-meta), 132.35 (s, SePh-ortho), 139.01 (s, C4), 155.86 (s, C2,6). 19F NMR (376.5 MHz, δ, ppm):–72.9 [s, 1J(FP) = 712.4 Hz, 6F, PF6]. 31P NMR (162 MHz, δ, ppm): −144.2 [hept, 1J(FP) = 712.3 Hz, 1P, PF6]. MS [ESI(+), m/z, %]: 481.8 [Cu(SeNSe)+ as base peak], 419.9 [SeNSe+ + H] [SeNSe = 2,6-(PhSeCH2)2C5H3N]. IR (ATR, cm−1): 1594 (w), 1568 (w) [ν(C = C)], 1478 (w), 1457 (m) [ν(C = N)], 828 (vs) [ν(PF6)].
4.2.7 [Cu{2,6-(PhSeCH2)2C5H3N}2](PF6) (8)
[Cu(MeCN)4]PF6 (0.036 g, 0.1 mmol) was added in one portion to a solution of 4 (0.082 g, 0.2 mmol) in anhydrous CH2Cl2 (40 mL). The reaction mixture was stirred at room temperature, under argon, for 2 h. The solvent was removed in vacuum. The remaining solid was washed with hexane to gave the title compound 8 as a yellow solid. Yield: 0.094 g (92%). 1H NMR (400 MHz, δ, ppm): 4.37 [s,br, 2J(HSe) = 13.1 Hz, 8H, CH2], 7.22 (m, 10H, H3,5 + SePh-ortho + meta), 7.31 (m, 2H, SePh-para), 7.73 [m, 3J(HH) 7.7 Hz, 1H, H4]. 13C NMR (100.6 MHz, δ, ppm): 36.93 [s, 1J(CSe) = 61.7 Hz, CH2], 123.94 (s, C3,5), 128.23 (s, SePh-ipso), 128.53 (s, SePh-para), 129.61 (s, SePh-meta), 132.43 (s, SePh-ortho), 139.28 (s, C4), 156.91 (s, C2,6). 19F NMR (376.5 MHz, δ, ppm): –73.0 [s, 1J(FP) = 712.7 Hz, 6F, PF6]. 31P NMR (162 MHz, δ, ppm):–144.2 [hept, 1J(FP) = 712.7 Hz, 1P, PF6]. MS [ESI(+), m/z, %]: 900.8 [Cu(SeNSe)2+], 481.8 [Cu(SeNSe)+], 419.9 [SeNSe+ + H, as base peak] [SeNSe = 2,6-(PhSeCH2)2C5H3N]. IR (ATR, cm−1): 1593 (w), 1566 (m) [ν(C = C)], 1477 (m), 1455 (m) [ν(C = N)], 828 (vs) [ν(PF6)].
4.3 X-ray structure determination
The details of the crystal structure determination and refinement for compound 2a are given in Table 2.
X-ray crystal data and structure refinement for compound 2a.
| Empirical formula | C21H24Ag4F12N3O9 |
| Formula weight | 1121.91 |
| T (K) | 297(2) |
| Crystal system | Triclinic |
| Space group | P-1 |
| a (Å) | 10.83(2) |
| b (Å) | 13.90(3) |
| c (Å) | 13.90(3) |
| α (°) | 79.27(2) |
| β (°) | 70.73(2) |
| γ (°) | 70.73(2) |
| Volume (Å3) | 1859(6) |
| Z | 2 |
| ρcalc (g cm−3) | 2.004 |
| Absorption coefficient (mm−1) | 2.180 |
| F(000) | 1078 |
| Crystal size (mm3) | 0.31 × 0.26 × 0.21 |
| Reflections collected | 13132 |
| Independent reflections | 6486 [Rint = 0.0343] |
| R1 (I > 2σ(I)) | 0.0616 |
| wR2 | 0.1256 |
| Goodness-of-fit on F2 | 1.092 |
Data were collected at room temperature on Bruker SMART APEX diffractometer, using graphite-monochromated Mo-Ka radiation (λ = 0.71073 Å). For this purpose the crystals were mounted on a cryoloop. The structures were refined with anisotropic thermal parameters. The hydrogen atoms were refined with a riding model and a mutual isotropic thermal parameter, except the one from the ethanol group which was not determined. The O(3) and O(4) atoms from a OOCCF3 fragment and the four CF3 groups are disordered over two positions and were refined with the following occupancies: 38:62, 37:63, 40:60, 60:40 and 39:61, respectively. For structure solving and refinement the software package SHELX-97 was used [26]. The drawings were created with the Diamond program [27].
Crystallographic data for the structural analysis of compound 2a have been deposited with the Cambridge Crystallographic Data Centre CCDC No. 865938. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; tel.; +44 1223 336 408; fax: +44 1223 336 033; deposit@ccdc.cam.ac.uk).
Acknowledgements
Financial support from the National University Research Council (CNCSIS, Romania; Research Project TD66/2007) and Ministerio de Economía y Competitividad (Spain, MEC/FEDER CTQ2010-20500-C02-01) is gratefully acknowledged. We also thank the National Center for X-ray Diffraction (Babes-Bolyai University, Cluj-Napoca, Romania) for support in the solid state structure determination.