

1 Introduction
While many structural types of single-molecule magnets, SMMs, are now known, spanning a wide range of metals, a majority of them are complexes containing the high spin MnIII ion which contributes a large spin and uniaxial anisotropy [1]. That notwithstanding, an approach that is increasingly gaining ground is the incorporation of lanthanide ions into SMM systems since these can contribute up to seven unpaired electrons and many show significant single-ion anisotropy. In particular, the creation of 3d-4f systems using lanthanides to modulate the magnetic properties of transition metal single-molecule magnets has become a very active area of research in recent years [2–5].
The interactions between 4f- or 4f- and 3d-electronic systems are essentially of magnetic dipolar nature and the contribution of the exchange interaction term appears to be very small, if not negligible, compared with that of the dipolar term in coordination clusters involving 4f ions. Furthermore, the mechanisms of slow relaxation of the magnetisation can be different for 3d and 4f ions. Nevertheless, it is clear that the resulting electronic structure, and therefore the magnetic behaviour, of coordination clusters built from both 3d and 4f ions can lead to SMMs with higher barriers Δeff to reversal of magnetization, and it is therefore of interest to investigate such clusters.
Earlier efforts in synthesising 3d/4f coordination clusters mostly concentrated on Cu/Gd systems which are usually ferromagnetically coupled, leading to high spin ground states but showing negligible anisotropy [6]. More recently, the idea of combining 3d and 4f metals ions so that one or both partners contribute high single-ion anisotropy as well as significant spin has been explored. For example, CuDy2 or CoII2Gd compounds were recently reported with Δeff around 47 to 28 K [2b,5]. Similarly, the combination of manganese or iron and lanthanides has resulted in barriers Δeff of up to 103 K [3n] and 33.4 K [4f], respectively.
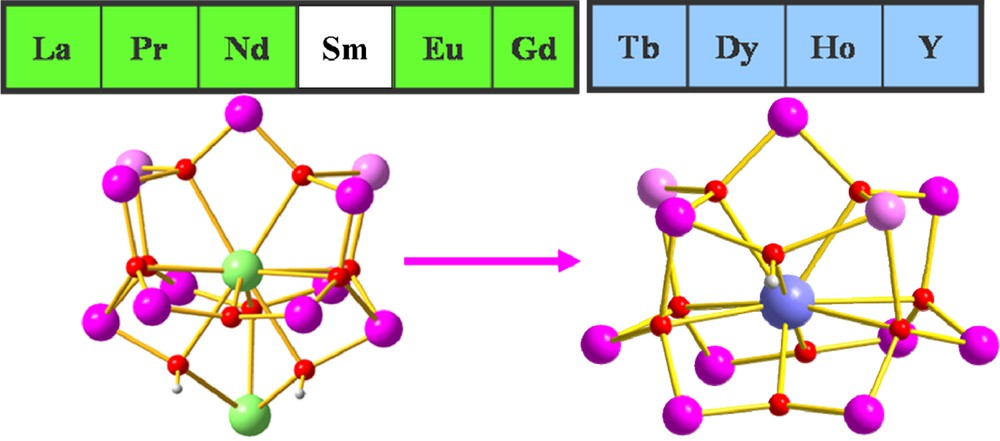
As part of intense efforts aiming to synthesise novel types of Mn-Ln compounds, we and several other groups have been exploring mixed-metal cluster complexes that have the appropriate properties to function as SMMs [3]. Our own contributions to this new field have included Mn11Gd2 [3d], Mn5Ln4 [3e], Mn10Ln2 [3f], Mn2Ln2 [3k], Mn2Ln3 [3j], Mn18Dy [3h] and Mn4Ln4 [3l]. Following on from our previous report on a {Mn11Gd2} complex [3d] which shows SMM properties, and the synthesis of a series of complexes with lighter lanthanides isostructural to the {Mn11Gd2} [7], we decided to apply the same synthetic procedure to the heavier lanthanides. Many reactions in various solvents were explored, but only after excluding furoic acid from the original reaction conditions and using diffusion of heptane into the reaction mixture could we isolate and characterise four isostructural dodecanuclear [MnIII9MnII2LnIII(O)8(OH)(piv)16(NO3)(CH3CN)]·xCH3CN·yC7H16 (piv = pivalate; x = ½, y = ¾, Ln = Tb (1); x = 2, y = ½, Ln = Dy (2), Ho (3), and Y (4)) complexes. Compounds 1-4 all exhibit SMM behaviour, showing out-of-phase signals in their ac susceptibilities; 1-3 additionally show hysteresis loops in microsquid measurements made below 1 K.
2 Experimental
2.1 General
All the reactions were carried out under aerobic conditions. [MnIII2MnII4O2(piv)10(4-Me-py)2.5(PivH)1.5] (2) was obtained as reported [3d]. Elemental analyses for C, H and N were carried out at the Institute for Inorganic Chemistry at Karlsruhe Institute of Technology.
2.2 Preparation
[Mn11Dy(O)8(OH)(piv)16(NO3)(CH3CN)]·2CH3CN·½C7H16 (2·2CH3CN·½C7H16): a stirred slurry of [Mn6O2(Piv)10(4-Me-py)2.5(PivH)1.5] (0.20 g, 0.11 mmol) in MeCN (10 ml) was heated to 70 °C, followed by addition of Dy(NO3)3·6H2O (0.20 g, 0.44 mmol), resulting in a dark-brown solution after 20 min. After stirring under reflux for an additional 30 min, the solution was cooled to room temperature and divided between five small (8 ml) vials, each with 2 ml of solution. Each of these vials was placed into a larger (25 ml) vial containing 2 ml of heptane and the larger vial was sealed. After 2–3 days some white microcrystalline powder appeared on the walls and bottom of the small vials. At this point each large vial was opened every day to check for monocrystals with a microscope and then resealed. After 7–8 days the white material disappeared and brown crystals began to appear. After a further 7 days the brown crystals were collected by filtration, washed with MeCN + H2O (1:1) to remove residual traces of the (water-soluble) white impurity, and dried in air. A similar yield of the final crystals was obtained after more than 4–5 weeks if the big vials were not opened during the crystallization period. With the procedure which is to briefly open the big vial every day, the crystallisation period can be shortened to 2 weeks. Probably, this procedure, in addition to faster evaporation of solvent, facilitates also the release of the pressure which may speed up the crystallisation. Combined yield from all vials = 0.065 g (46.0% based on Mn). Calc. for C82.1H149.8DyMn11NO44 (= 2·0.3 C7H16): C 37.61, H 5.76, N 0.53%; found: C 37.47, H 5.82, N 0.47%. IR (KBr disk, cm−1): 3430 (s), 2965 (s), 2932 (s), 2908 (s), 2323 (w), 1554 (vs), 1485 (vs), 1461 (s), 1431 (vs), 1382 (s), 1360 (s), 1303 (s), 1230 (vs), 1032 (w), 939 (m), 893 (m), 800 (s), 785 (m), 665 (s), 623 (s), 587 (s), 458 (w).
[Mn11Tb(O)8(OH)(piv)16(NO3)(CH3CN)]·½CH3CN·¾C7H16 (1·½CH3CN·¾C7H16): this compound as brown crystals was obtained using the same procedure as for 2 with Tb(NO3)3·6H2O in place of Dy(NO3)3·6H2O. Total yield: 0.060 g (43.0% based on Mn). Calc. for C82.1H149.8TbMn11NO44 (= 1·0.3 C7H16): C 37.66, H 5.77, N 0.53%; found: C 37.48, H 5.79, N 0.47%. IR (KBr disk, cm−1): 3431 (s), 2964 (s), 2932 (s), 2907 (s), 2323 (w), 1553 (vs), 1485 (vs), 1461 (s), 1431 (vs), 1381 (s), 1359 (s), 1302 (s), 1229 (vs), 1032 (w), 938 (m), 893 (m), 799 (s), 785 (m), 664 (s), 623 (s), 587 (s), 457 (w).
[Mn11Ho(O)8(OH)(piv)16(NO3)(CH3CN)]·2CH3CN·½ C7H16 (3·2CH3CN·½C7H16): this compound as brown crystals was obtained using the same procedure as for 2 with Ho(NO3)3·6H2O in place of Dy(NO3)3·6H2O. Yield: ∼0.055 g (39.0% based on Mn). Calc. for C81.4H148.2HoMn11NO44 (= 3·0.2 C7H16): C 37.40, H 5.71, N 0.53%; found: C 37.23, H 5.78, N 0.46%. IR (KBr disk, cm−1): 3419 (s), 2964 (s), 2932 (s), 2907 (s), 2320 (w), 1553 (vs), 1485 (vs), 1460 (s), 1430 (vs), 1381 (s), 1359 (s), 1302 (s), 1228 (vs), 1032 (w), 938 (m), 893 (m), 799 (s), 785 (m), 667 (s), 623 (s), 587 (s), 457 (w).
[Mn11Y(O)8(OH)(piv)16(NO3)(CH3CN)]·2CH3CN·½ C7H16 (4·2CH3CN·½C7H16): this compound as brown crystals was obtained using the same procedure as for 2 with Y(NO3)3·6H2O in place of Dy(NO3)3·6H2O. Yield: ∼0.050 g (35.7% based on Mn). Calcd for C82.1H149.8YMn11NO44 (= 4·0.3 C7H16): C 38.70, H 5.92, N 0.55%; found: C 38.51, H 6.11, N 0.46%. IR (KBr disk, cm−1): 3412 (s), 2965 (s), 2932 (s), 2907 (s), 2319 (w), 1551 (vs), 1486 (vs), 1460 (s), 1431 (vs), 1379 (s), 1359 (s), 1302 (s), 1229 (vs), 1033 (w), 938 (m), 892 (m), 797 (s), 782 (m), 663 (s), 620 (s), 585 (s), 458 (w).
2.3 Magnetic measurements
The magnetic susceptibility measurements were carried out on polycrystalline samples using a Quantum Design SQUID magnetometer MPMS XL over the temperature range 1.8–300 K for dc applied fields up to 7 T. Alternating current (ac) susceptibility measurements were performed with an oscillating field of 3 Oe and ac frequencies ranging from 1 to 1500 Hz. M versus H measurements were performed at 100 K to check for the presence of ferromagnetic impurities and none were found. The magnetic data were corrected for the sample holder and the diamagnetic contribution.
2.4 Crystal structure determinations
Data for compounds 1-3 were collected at 100 K on a Bruker SMART Apex CCD diffractometer using graphite-monochromated MoKα radiation. Semiempirical absorption corrections were made using SADABS [8a]. The structures were solved using direct methods, followed by full-matrix least-squares refinement against F2 (all data) using SHELXTL [8b]. Anisotropic refinement was used for all ordered non-H atoms; organic H atoms were placed in calculated positions. Disordered t-Bu groups were refined with isotropic partial methyl carbon atoms, using geometrical restraints. One disordered lattice MeCN in the lattice of compounds 1 and 2 could not be refined satisfactorily and was handled using the SQUEEZE option in PLATON [9]. Lattice heptane molecules were disordered over special positions, and were refined with half-occupancy C atoms with geometrical similarity restraints. Crystals of 4 diffracted with rather low intensity; however their unit cell showed that they were isotypic to 2 and 3. Since the crystals can lose lattice solvent rather readily on exposure to air, the compositions and formula weights of the samples used in the magnetic measurements were estimated from microanalytical data, particularly when these came from different batches from those used for the structure determinations.
Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 862345-862347. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK: http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi, data_request@ccdc.cam.ac.uk, or fax: +44 1223 336033.
3 Results and discussion
3.1 Structural analysis
The complexes were structurally characterized using single crystal X-ray crystallography. The details of crystallographic data and structure refinement parameters are summarized in Table 1. Compounds 2-4 crystallize isotypically in the monoclinic space group P21/n with Z = 4, compound 1 in P21/c with Z = 4. Nonetheless, the molecular structure of the Mn11Tb complex in 1 is very similar to those in 2 and 3, both in terms of atomic connectivity and the orientation of the MnIII Jahn-Teller elongation axes.
Crystallographic data for compounds 1-4.
| 1 | 2 | 3 | 4a | |
| Formula | C88.25H161.5Mn11N2.5O44Tb | C89.50H162DyMn11N4O44 | C89.5H162HoMn11N4O44 | - |
| Mr | 2724.96 | 2765.07 | 2767.50 | |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P 21/c | P 21/n | P 21/n | P 21/n |
| a [Å] | 28.531(2) | 16.5698(11) | 16.5879(10) | 16.559(2) |
| b [Å] | 16.4130(12) | 26.9753(17) | 26.9126(16) | 26.933(3) |
| c [Å] | 28.080(2) | 27.2615(18) | 27.2793(16) | 27.213(3) |
| β [°] | 110.114(1) | 91.142(1) | 91.258(1) | 91.134(3) |
| V [Å3] | 12347.3(16) | 12182.8(14) | 12175.2(13) | 12133(4) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcd [g/cm−3] | 1.466 | 1.508 | 1.510 | - |
| μ(Mo-Kα) [mm−1] | 1.724 | 1.781 | 1.819 | - |
| F(000) | 5602 | 5680 | 5684 | - |
| T/K | 100(2) | 100(2) | 100(2) | 100(2) |
| Collected reflections | 88557 | 89875 | 82809 | - |
| Unique reflections | 27794 | 26559 | 27628 | - |
| Rint | 0.0708 | 0.0332 | 0.0385 | - |
| Reflns with I > 2σ(I) | 21917 | 22807 | 24237 | - |
| Refined parameters | 1355 | 1330 | 1327 | - |
| R1 [I > 2σ(I)] | 0.0567 | 0.0320 | 0.0428 | - |
| S (all data) | 1.110 | 1.001 | 1.116 | - |
| wR2 (all data) | 0.1195 | 0.0785 | 0.0935 | - |
a The structure of 4 could not be fully refined, as a result of poor crystal quality.
The structure of the compound 2 will be discussed in detail as a representative example (Fig. 1). The oxidation states of the Mn centres were determined by bond-valence sum (BVS) calculations [10], with Mn(1), Mn(2) and Mn(5)-Mn(11) assigned as MnIII, and Mn(3) and Mn(4) as MnII. The Mn centres all have octahedral coordination geometries except for Mn(11), which is square-pyramidal. The high spin MnIII ions all show the expected Jahn-Teller elongations. The assignments of O(1)-O(6) as (μ4-O)2−, O(8) and O(9) as (μ3-O)2−, and of O(7) as (μ3-OH)− were also confirmed by BVS calculations and the H-atom on O(7) could be located and refined.
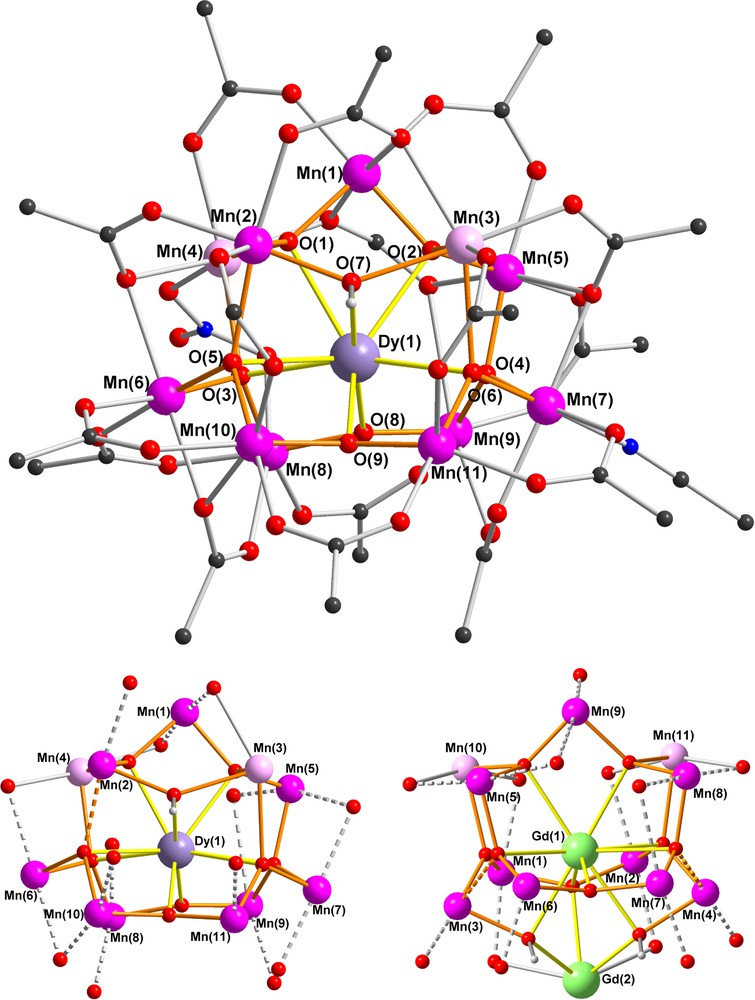
Structure of complex 2 (above); the arrangement of the nine MnIII Jahn-Teller (JT) elongation axes (dotted lines) in 2 (below left) and in the Mn11Gd2 complex of [3d] (below right). (MnII pink, MnIII purple, Dy lilac, Gd green, N blue, O red, C black, methyl groups of pivalates omitted for clarity.)
The Mn11Dy core of 2 is closely related to that of our previously reported “bell-shaped” Mn11Ln2 complexes [3d,7] and its core is compared with that of the Mn11Gd2 compound in Fig. 1. The MnIII and MnII centres again form the shell of the bell with Mn(1) at the apex, Mn(2)-Mn(5) at the shoulder of the bell, and the remaining six MnIII centres forming the rim of the bell. Although the two MnII centres in 2 are once again located on the shoulder of the bell, they are now on opposite sides of the bell, whereas in the Mn11Gd2 compound they were on the same side and linked by a pivalate bridge. Dy(1) is nine-coordinate and is connected to each of the eleven Mn centres through the six (μ4-O), the two (μ3-O) and the (μ3-OH) bridges to give a rather irregular coordination polyhedron (Fig. 2). The Mn11Dy core can thus be considered as derived from the “bell-shaped” Mn11Ln2 core [3d,7], but with the second Dy at the end of the bell's “clapper” cut off (Scheme 1).
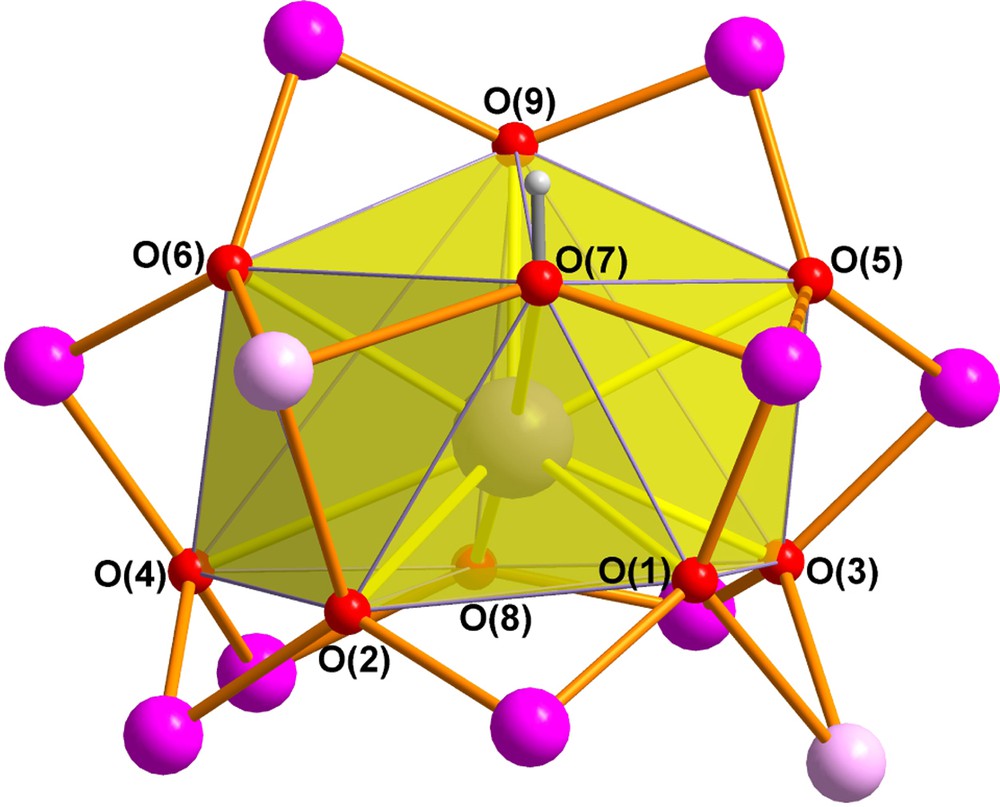
The (O)8(OH) coordination polyhedron of Dy(1) in 2.
Of the sixteen pivalate ligands, the unusual (μ4,η2,η2) bridging mode, which was seen in the Mn11Ln2 compounds [3d,7], is observed for one pivalate ligand. Five further pivalates adopt a (μ3,η2,η1) triply-bridging mode, while the remaining pivalates and a NO3− anion form simple syn,syn-bridges between pairs of metal centres. The octahedral coordination of Mn(7) is completed by a coordinated acetonitrile molecule. The molecules are well separated within the crystal structure and no intermolecular hydrogen bonds were observed in the crystal packing.
3.2 Magnetic properties
The dc magnetic susceptibility data for compounds 1-4 were collected in the 1.8–300 K temperature range under a field of 0.1 T (Fig. 3). The dc magnetic data, together with the Weiss constants obtained from Curie-Weiss plots using the data above 130 K, are summarized in Table 2. The χT vs T plots for all compounds show similar profiles with the χT product decreasing in a monotonic fashion over the full temperature range indicating the presence of dominant antiferromagnetic (AF) interactions. The room temperature χT value of 30.06 cm3 K mol−1 in 4 is lower than the expected value for nine MnIII and two MnII non-interacting ions (35.75 cm3 K mol−1), suggesting that the overall AF interactions present between the constituent Mn ions are relatively strong. This is supported by the relatively large and negative value for the Weiss constant of −50.9 K (Table 2) and also by the small magnitude of the magnetization at 2 K under a 7 T field, 7.0 μB, which is significantly lower than that (46 μB) calculated for a parallel alignment of all the spins (Fig. S1). Furthermore, extrapolation of the in-phase susceptibility product (χ’T) of 4 at very low temperature approaches 2.4 cm3 K mol−1 (Fig. S2), suggesting a small spin ground state of S ≈ 2 in this compound.
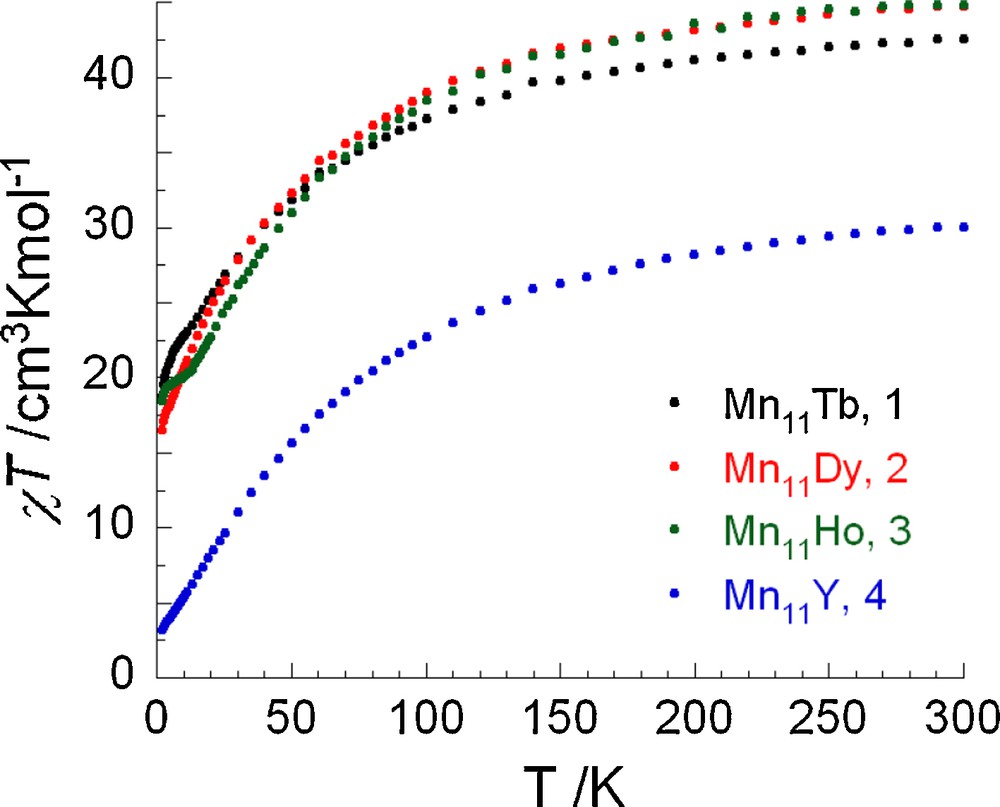
χT vs T plots for complexes 1-4 (Mn11Tb, Mn11Dy, Mn11Ho, and Mn11Y, respectively) in 0.1 T applied dc field.
Ground state and room temperature χT values for Mn11Ln core.
| Compounds | Ground state of Ln3+ | Curie constant for Ln3+ | Predicted χT (cm3mol−1K) | Observed χT (cm3mol−1K) | Weiss constant θb (K) |
| Mn11Tb (1) | 7F6 | 11.82 | 41.88a | 42.60 | −22.6 |
| Mn11Dy (2) | 6H15/2 | 14.17 | 44.23a | 44.79 | −22.8 |
| Mn11Ho (3) | 5I8 | 14.07 | 44.13a | 44.88 | −26.6 |
| Mn11Y (4) | Diamagnetic | 0 | 35.75 | 30.06 | −50.9 |
a The χT product of the Mn11 unit is used as the experimental value of the Y analogue, 4.
b θ is obtained by fitting the Curie-Weiss law from 130–300 K.
For compounds 1–3, the experimental χT product observed at 300 K is in good agreement with the sum of the value of 4 (30.06 cm3 K mol−1) and the Curie constant of single TbIII, DyIII and HoIII ions, respectively (Table 2). In order to assess the interaction between the paramagnetic LnIII and the Mn11 unit, we can subtract the molar susceptibility contribution of the YIII analogue from the susceptibilities of 1-3. The ‘subtracted’ χT product increases with decreasing temperature (Fig. S3) suggesting that the single LnIII ion is ferromagnetically coupled to the Mn11 unit. This is in line with the fact that the Weiss constants of 1-3 are much less negative than that of the diamagnetic YIII analogue 1. The ferromagnetic interaction between LnIII and the Mn11 unit is undoubtedly of smaller magnitude than the AF interactions amongst the MnII/III ions in the shell.
The field dependence of the magnetization at low temperatures for each of the compounds 1-3 shows a relatively fast increase of the magnetization at low fields, followed by an almost linear increase without clear saturation up to 7 T, where it reaches 14.8, 14.5, and 12.3 μB for 1, 2 and 3, respectively (Fig. S1). This observation is indicative of the presence of magnetic anisotropy and/or excited levels mixing into the ground state.
As a result of the magnetic anisotropy present in these compounds, the ac susceptibility measurements were checked under zero dc field. There is only a very weak out-of-phase signal shown above 1.8 K for compound 4, while for 1, 2 and 3 out-of-phase signals are clearly detected below 3 K (Fig. S4). The frequency dependent out-of-phase signals suggest that all these compounds exhibit slow relaxation of its magnetization. The weak slow relaxation observed in compound 4 results from the small contribution of the spin ground state (S ≈ 2) arising from the strong AF couplings between the Mn ions. The significant enhancement to the relaxation observed on incorporation of the 4f ions in place of the YIII is similar to what we observed for the Mn4Ln5 compounds [3e].
In order to confirm SMM behaviour present in these compounds, single crystal magnetization measurements were performed using an array of micro-SQUIDs at temperatures down to 40 mK [11]. Below 1.0 K hysteresis loops are seen in magnetization vs field studies whose coercivities increase with decreasing temperature and increasing sweeping rate (Fig. 4 and Fig. S5). The shape and the width of hysteresis loops observed for the Mn11Tb (1) and Mn11Dy (2) compounds are rather similar in that the coercivity remains narrow at low fields indicating fast tunnelling occurs at H = 0, but the Mn11Ho compound 3 has a somewhat larger coercivity suggesting less tunnelling at H = 0 in this case. The tunnelling at H = 0 present in the Mn11Tb (1) and Mn11Dy (2) compounds is strong enough to make it impossible to obtain a reliable Arrhenius plot, even though slow relaxation is seen. However, for the Mn11Ho compound 3, the dc magnetization decay could be monitored with time, with the resulting effective energy barrier and the relaxation time of 6.0 K and 8.6 × 10−4 s, respectively (Fig. 5). Since there are no obvious pathways for intermolecular interactions the observed hysteresis results from intramolecular slow relaxation of the magnetization.
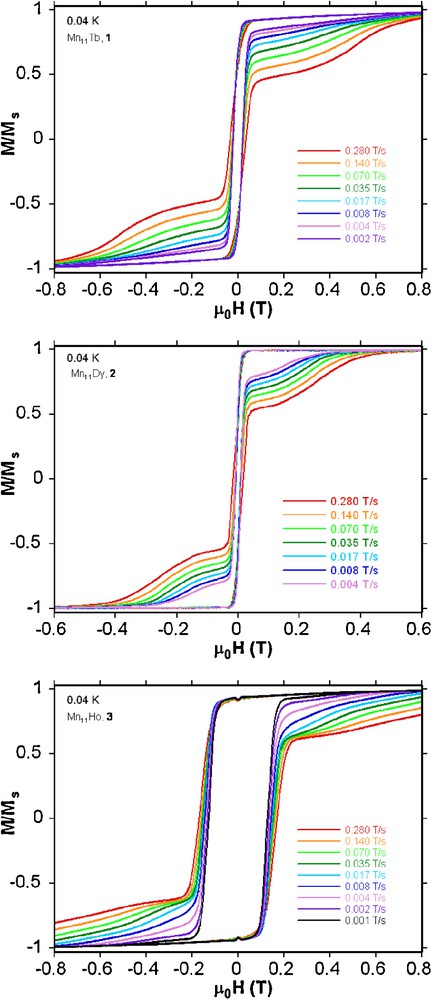
Magnetization (M) versus applied DC field (H) for single crystals of compounds 1-3 at 0.04 K with different sweeping rates. The magnetization is normalized to its saturation value Ms.
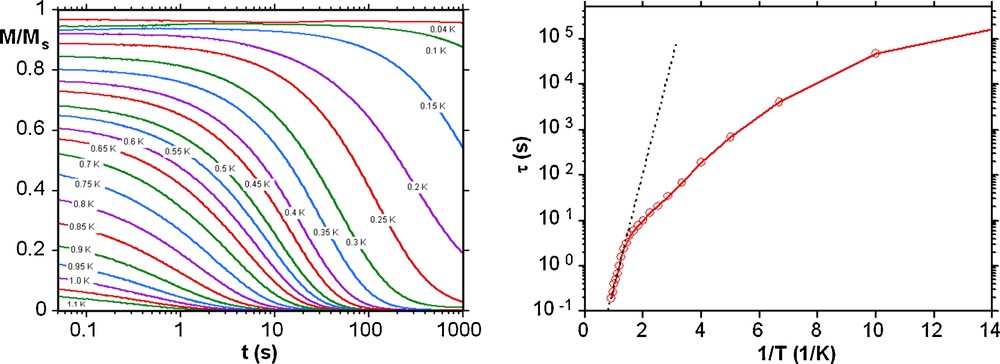
Left: magnetization (M) versus time decay plots in zero dc field for a single crystal of 3; the magnetization is normalized to its saturation value Ms. Right: Arrhenius plot of the relaxation time (τ) versus 1/T of 3 using the dc magnetization decay data. The dashed line is the fit of data in the thermally activated region to the Arrhenius equation; see the text for the fit parameters.
The Mn11Y compound 4 and our previously reported Mn11La2 complex [7] have similar bell-shaped MnIII9MnII2 cores and their rare-earth ions are diamagnetic. Although they both have small spin ground states with S ≈ 2, 4 shows weak out-of-phase signals in its ac susceptibility, whereas the Mn11La2 does not. These two compounds are respectively isostructural to 2 and to the Mn11Gd2 complex, for which the Mn11 cores and orientations of the MnIII Jahn-Teller axes are compared in Fig. 1. Although quantitative analysis of the Jahn-Teller axes is not possible, comparison of the two cores in Fig. 1 suggests that these axes might be better aligned in the Mn11Ln series than in the Mn11Ln2 one. In the Mn11Ln compounds (Fig. 1, below left), seven of the Jahn-Teller axes are aligned approximately in the direction of the Ln(1)-Mn(1) vector, with only those of Mn(1) and Mn(5) orientated perpendicular to this direction. In the Mn11Ln2 series (Fig. 1, below right) three such axes, those for Mn(5), Mn(8) and Mn(9), are oriented essentially perpendicular to the other six. This would be in line with the idea that compound 4 should have a larger overall molecular uniaxial anisotropy arising from the tensorial sum of the anisotropies of the individual ions.
It is appropriate at this point to re-examine the coordination polyhedron of the DyIII ion in 2, which is noticeably oblate, with Dy(1) lying rather close to the plane of the pentagon defined by the oxo ligands O(1), O(2), O(3), O(4) and O(8). In their review of lanthanide anisotropy and single-molecule magnet behaviour, Rinehart and Long point out that such a coordination geometry is rather unusual for DyIII, with axially-dominated geometries being more common. Such prolate geometries result in uniaxial single-ion anisotropies for ions such as TbIII and DyIII, for which the MJ states with high |J| have oblate electron densities [12]. The coordination polyhedra of the lanthanides in 1 and 2 are therefore unlikely to favour uniaxial single-ion anisotropy for their TbIII or DyIII ions. Consequently, it is likely that the enhanced SMM behaviour observed for 1 and 2 compared with 4 mainly result from the ferromagnetic coupling of the spins on the TbIII or DyIII to the anisotropic Mn11 shells, just as was found for the Mn11Ln2 compounds [3d,7], rather than from the LnIII ions making any significant single-ion uniaxial anisotropy to the molecules. The angular electron density distribution for the HoIII MJ levels with high |J| are such that they are expected to be less destabilised than for TbIII and DyIII [12], which might be predicted to improve SMM behaviour for 3 compared to 1 and 2, as is, in fact the case here. Indeed, the oblate coordination polyhedron found for the LnIII ions in 1-3 is more likely to promote uniaxial single-ion anisotropy for ions such as ErIII or YbIII [12], but unfortunately it was not possible to isolate such Mn11Ln compounds with lanthanides heavier than holmium.
It is noticeable that no well-resolved steps in the hysteresis loops of compounds 1-3 due to resonant quantum tunnelling of magnetization (QTM) were observed. In a recent report of a Mn12Gd complex, which also has a central lanthanide surrounded by a shell of manganese ions, it was proposed that the number of O2− ions bridging between the manganese and the lanthanide ion(s) in such MnxLny clusters plays a crucial factor in determining whether well-resolved quantum tunnelling behaviour is observed or not, and that eight such oxo bridges were necessary to observe steps in the hysteresis [3g]. However, in compounds 1-3 we do indeed have eight O2− ions bridging between the manganese centres and the lanthanide ions, but no clear QTM steps can be observed in the hysteresis loops, in contradiction of the perhaps overly simplistic proposal in [3g]. It is unlikely that the number of oxo bridges is the only factor contributing to the presence of quantum tunnelling. The precise structural topologies, the relative orientations of the Jahn-Teller anisotropies of the MnIII centres, differences in the Mn-O-Mn and Mn-O-Ln angles, and the specific anisotropy brought to the molecule by the rare-earth ion(s) will all influence any quantum behaviour in such compounds.
4 Conclusions
In summary, a family of dodecanuclear heterometallic Mn11Ln complexes with one lanthanide ion encapsulated within a large manganese shell has been prepared. All these complexes show magnetic slow relaxation indicating that they are SMMs. The SMM behaviour of 1-3 was confirmed by micro-SQUID measurements, with well-resolved hysteresis loops below 1.0 K. Comparison with the Mn11Y analogue 4 suggests that the enhancement of SMM behaviour observed in 1-3 most likely results from the additional spin on the coordination cluster resulting from the ferromagnetic coupling of the incorporated TbIII, DyIII or HoIII ions to the anisotropic but rather low-spin (S ≈ 2) Mn11 shell, since the local coordination geometries of the individual LnIII ions are considered unlikely to promote uniaxial single-ion anisotropy.
Acknowledgements
This work was supported by the DFG (CFN) and AvH Foundation (V.M.).