

1 Introduction
Oxygenated cage structures are frequently isolated from natural sources. Some famous examples have inspired beautiful total syntheses such as FR182877 (1) [1], phomoidride B (2) [2], trichodimerol (3) [3], or ginkgolide (4) [4] (Fig. 1). The biosynthetic origin of these compounds usually involves highly oxidative biochemical processes followed by various types of cyclizations. Among them, the opening of epoxides is a powerful strategy, in general following Baldwin's rules [5]. Harringtonolide (5) [6] and tetrodecamycin (6) [7] are likely to follow such processes from transient cyclohexene epoxides during their biological formation.
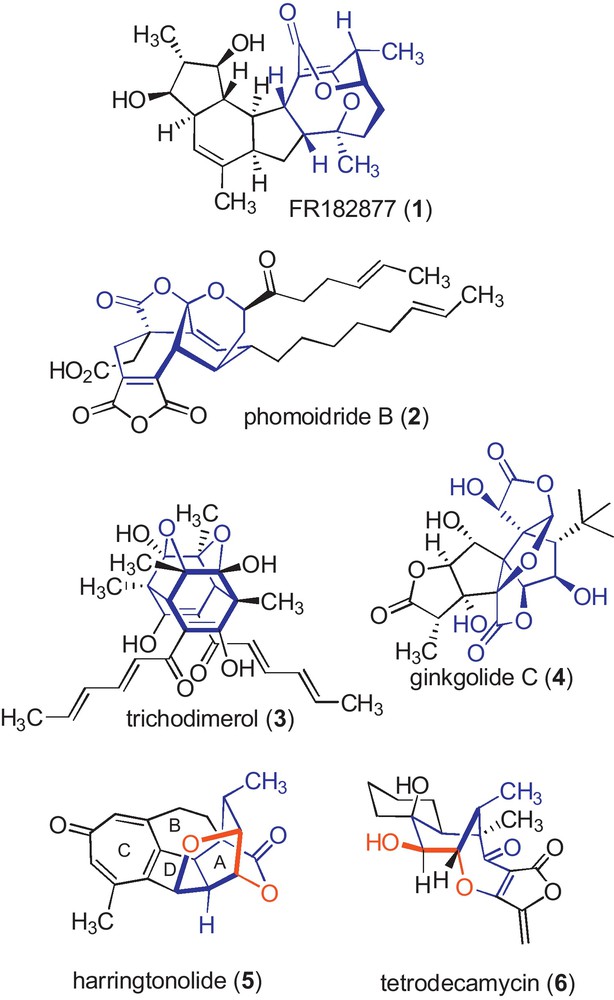
Examples of natural oxygenated cage architectures (in blue) and trans-dioxy structures possibly arising from opening of an epoxide in nature (in red).
During our work on the total synthesis of 5, we studied the reactivity of specific cyclohexene epoxides toward acid-catalyzed cyclization in order to generate the oxygenated cage structure of the natural product [8,9]. Monocyclic and bicyclic model compounds (circled structure in Scheme 1) were designed with a hydroxylated chain, which may participate in the formation of an oxetane (in accordance with the Baldwin rules) [5] or a tetrahydrofuran ring. Additionally, an ester was present on the cyclohexane ring in such a position that the formation of either a 5-membered or a 6-membered lactone may be possible after opening the epoxide. With such a functional triad, we were expecting cascade reactions to happen in order to form a cycloether and a lactone in a single process.
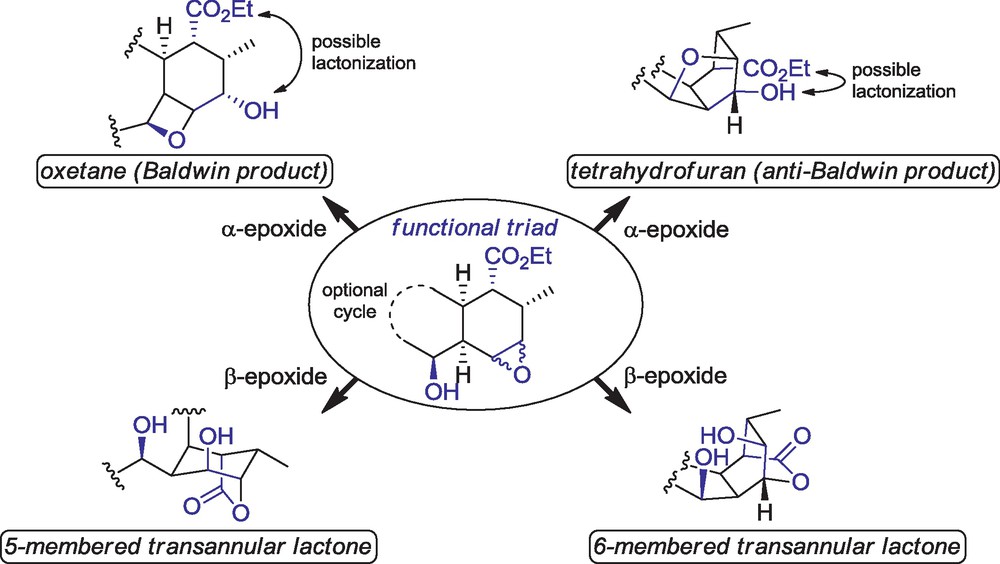
The functional triad toward acid-catalyzed cyclization events.
2 Synthesis of the hydroxy-epoxy-ester functional triads
The triads were synthesized from the advanced intermediate 7 derived by a stereodirected intramolecular Diels-Alder reaction already described in previous reports [10]. The intermediate 7 was treated in TFA/H2O at 80 °C (Scheme 2), leading to acetal hydrolysis and concomitant contraction of the lactone ring. From the diol product 8, both monocyclic and bicyclic systems of our study were constructed.
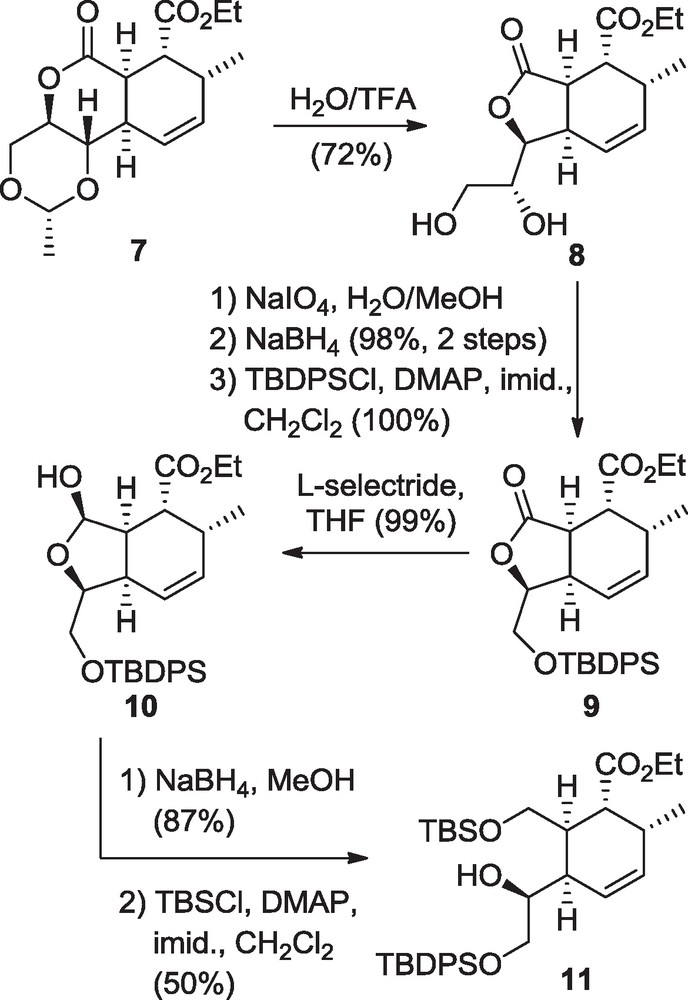
Synthesis of monocyclic branched cyclohexenes.
The branched monocyclic systems were built after the oxidative cleavage of diol 8 in the presence of sodium periodate, directly followed by aldehyde reduction and TBDPS-protection of the resulting primary alcohol, affording bicyclic compounds 9 (Scheme 2) [8]. L-Selectride reduction of the lactone provided the corresponding lactol in 99% yield. Unfortunately all attempts for Wittig methylenation of the 5-membered lactol 10 only resulted in unwanted side reactions or degradation. Instead, lactol reduction into the corresponding diol was undertaken in the presence of NaBH4 in MeOH, giving the expected products 11 after protection of the primary alcohol as TBS ether.
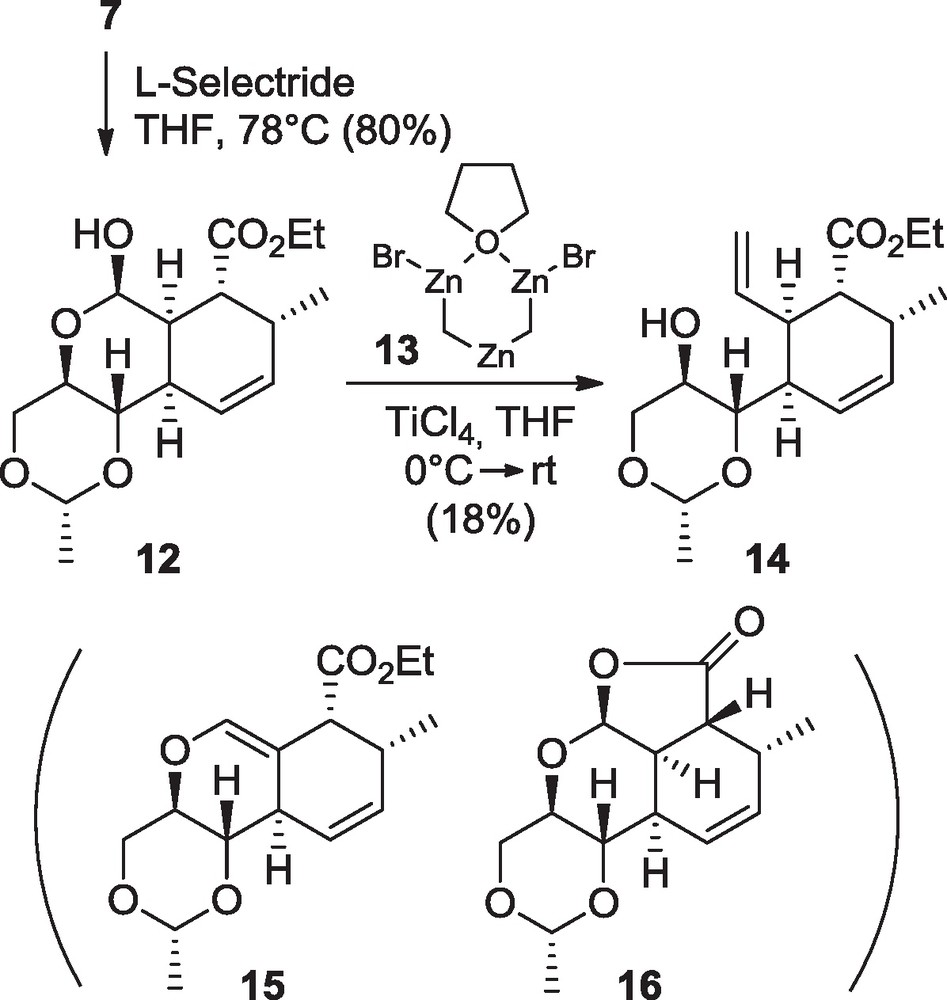
During this work, considerable attention was given to the Wittig methylation that was not possible on the 5-membered lactols like 10. Similarly the 6-membered lactol 12 derived by the cycloadduct 7 proved to be inert under the same reaction conditions (Ph3PCH3Br, NaHMDS, THF, –78 °C to rt). Nevertheless the use of the Nysted reagent 13 ((BrZnCH2)2Zn•THF) [11] in the presence of TiCl4 in THF at 0 °C afforded the desired substrate 14 in 18% yield, occasionally accompanied by compounds 15 and/or 16 when altering the conditions (Scheme 3). Even if this yield is far from being satisfactory, this is the first time, to the best of our knowledge, that an example of lactol methylation using the Nysted reagent is reported so far. Alternatively, with the aim of constructing the AD bicyclic system2 (Fig. 1) of harringtonolide 5 [9], the diol 8 was converted into the alkene 17 in 85% yield (Scheme 4) through an Appel reaction using the Garegg and Samuelsson conditions (PPh3, I2, imidazole, toluene, reflux) [12]. Gratefully with such a substrate, after L-selectride reduction of the lactone ring in 91% yield, the Wittig olefination was successfully undertaken. Only the semistabilized ylide Ph3P = CHPh (two equivalents) was found to give satisfactory yield with up to 96% of the diene 18. This was obtained after careful optimization to avoid the formation of by-product 21 possibly resulting from a poisonous combination of high basicity and inappropriate concentration of the medium.
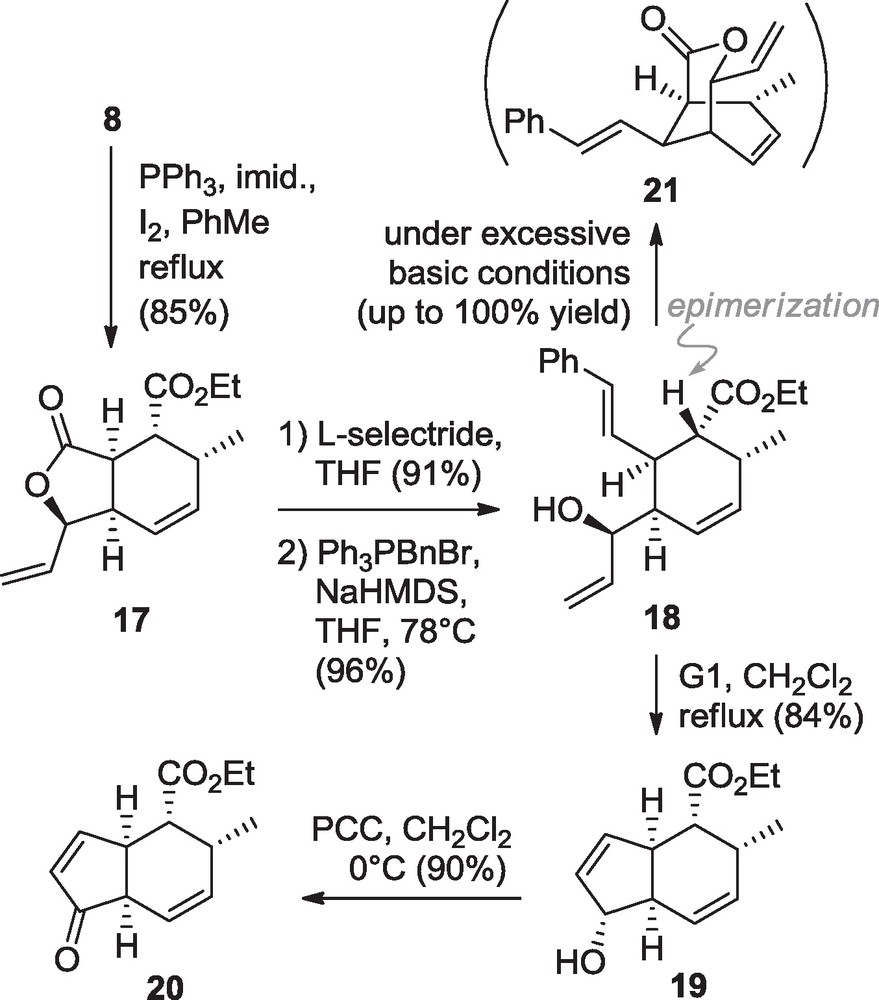
Synthesis of the bicyclic systems toward bicycle AD of harringtonolide 5.
Diene 18 was finally subjected to a ring-closing metathesis in the presence of Grubbs’ catalysts (either first or second generation, at reflux or room temperature in dichloromethane, 84% and 93% yields, respectively), providing the bicyclic allylic alcohol 19. After PCC oxidation, an electron-deficient enone 20 was obtained and this olefin was thus protected from the following epoxidation events.
During this work, additional cyclohexene systems were obtained as represented in Fig. 2. All were submitted to epoxidation conditions and their functional triad to cascade cyclization attempts. In fact, the geometry of the monocyclic compounds was not initially expected to be important in the selectivity of the epoxidation as these cyclohexenes are holding a pseudo-C2 symmetry axis. In these cases the nature of the R group (Box in Fig. 2) may influence the reaction. On the contrary, in the rigid bicyclic systems (19-20), the geometry may be determinant in the stereoselectivity of the epoxidation.
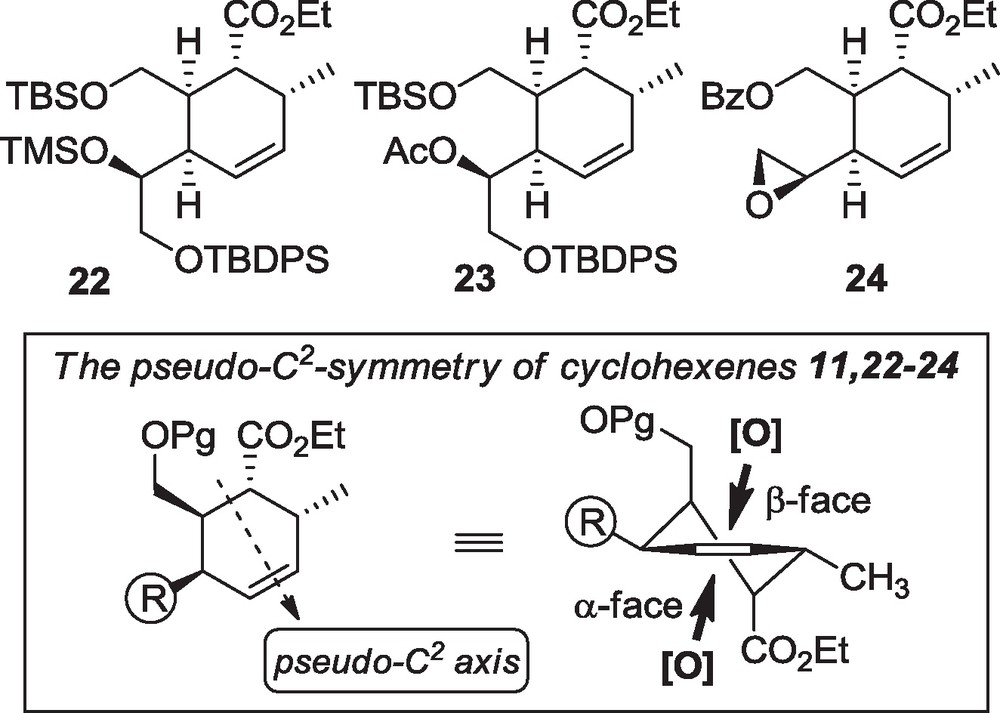
Functional group variations and geometry analysis (box) of monocyclic systems 11 and 22-24.
3 Epoxydation of selected monocyclic and bicyclic systems
Epoxidations attempts were engaged on the six subtrates 11, 19, 20, and 22–24. In many cases, mCPBA (buffered or not with NaHCO3) was not suited for these sensitive substrates and DMDO [13] was preferred. Except for the alcohol 11, poor selectivity was observed with the branched monocyclic cyclohexenes 22–24 (Table 1, entries 1–4), which was explained by the above-mentioned pseudo-C2 symmetry axis. In general, the β-epoxide was yet slightly favored, even with the use of a bulky protection on the hydroxyl group of the side chain R2 (Table 1, entry 2). With the alcohol 11, exclusive epoxidation on the β-face was observed, suggesting that the free hydroxyl group has an important directing effect by possible hydrogen binding with DMDO [14]. Surprisingly, with the bicyclic compound 20, only the β-epoxide (29b) was observed (entry 5). We suspected similar selectivity with substrate 19, but structure analysis of compound 30b was uncertain at this stage of the work (entry 6). In fact, after NaBH4 reduction of the enone on 29b (see compound 38 below), X-ray crystallography revealed the pseudo-axial conformation of the methyl group, which could thus represent a steric hindrance of the α-face for DMDO. Yet additional stereoelectronic effects are not excluded to explain this unexpected selectivity.
Results of the epoxydation of subtrates 11, 19, 20, and 22–24.
| Compounds | Yields (%) | Products Selectivity α:β | |
| 1 | 100 | ||
| 0:1 (25a/25b) | |||
| 2 | 91 | ||
| 2:3 (26a/26b) | |||
| 3 | 100 | ||
| 1:1 (27a/27b) | |||
| 4 | 90 | ||
| 2:5 (28a/28b) | |||
| 5 | 100a | ||
| 0:1 (29a/29b) | |||
| 6 | 100 | ||
| 0:1:0:0 (30a/30b/30c/30d) |
a Crude extract.
4 Rearrangement of the epoxide substrates under acid-catalyzed conditions
Attempts to functionalize monoepoxide 24, especially by opening with an acetylide in the presence of boron trifluoride etherate, led to unexpected reactivity (Scheme 5) [8]. Indeed the alkyne was not introduced but a complex rearrangement of 24 occurred, providing the cage structure 33. The mechanism would involve a set of secondary cations (31, 32) resulting from the opening of the oxirane in the presence of BF3, followed by migration of the cation through olefin switch and cyclopropane formation. The last cation would be intramolecularly quenched by the boron alkoxide to give the tetrahydrofuran cycle.
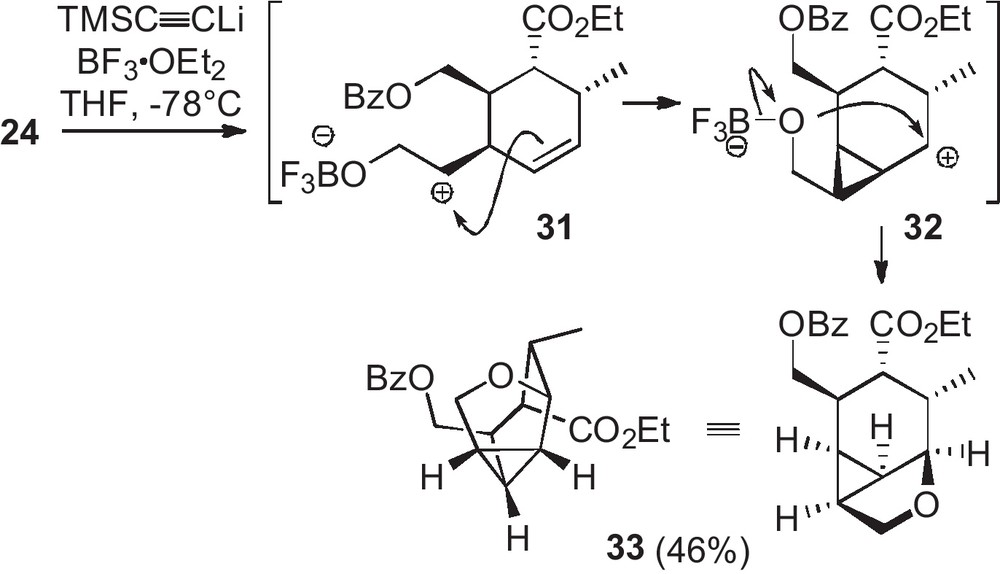
Unexpected reactivity of the epoxide 24 in the presence of BF3•OEt2.
Cascade cyclizations of several cyclohexene epoxides were undertaken under various acid-catalysis conditions3. The outcome of the reaction could be predicted by the Baldwin rules with no exception [5]. The diepoxide 28a was unable to react in cascade cyclization toward a polyether compound (Scheme 6). However, under acidic KBr pressure, the primary epoxide was opened and furnished the bromohydrine 34 isolated in 94% yield. It underwent cyclization into the oxetane 35 in 25% isolated yield in the presence of PTSA in toluene at 80 °C, as a result of 4-exo-tet cyclization. In these conditions, no further cyclization, especially toward a 5-membered lactone, was observed.
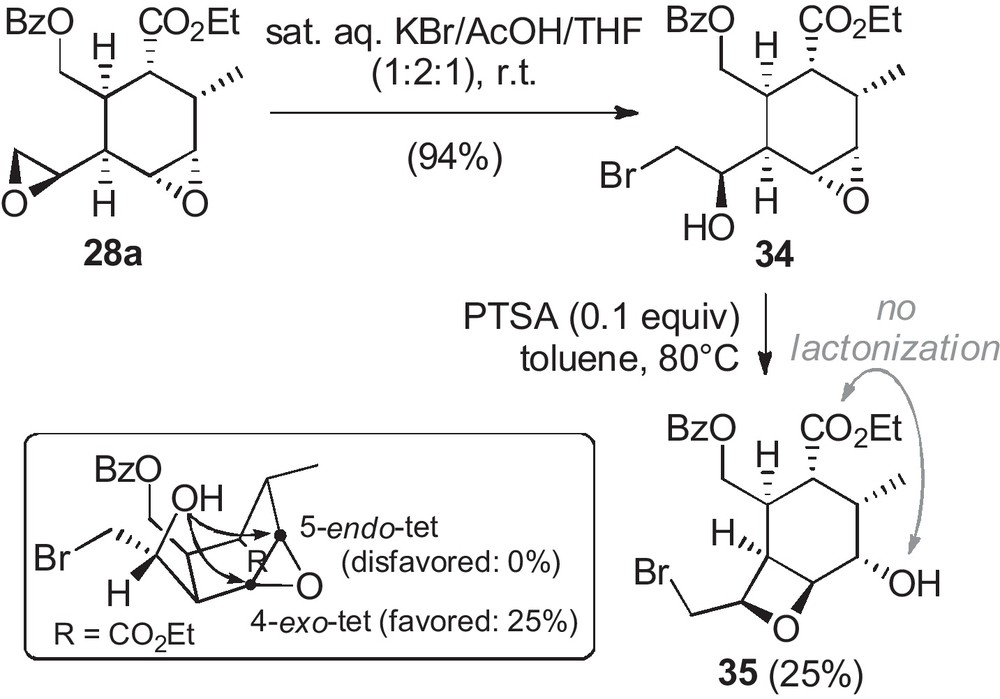
Reactivity of the diepoxide 28a under acidic conditions.
The formation of a lactone cycle (37) was, however, observed from the β-epoxide 25b under the same reaction conditions (Scheme 7). In fact, since no intramolecular cyclization was expected with the stereochemistry of 25b, regioselective opening of the epoxide could only occur in the presence of residual water, leading to the formation of the corresponding triol 36 isolated in 46% yield. During this reaction, a transannular cyclization of the α-hydroxyl group with the α-ester furnished the bridged 5-membered lactone 37 isolated in 25% yield. The triol 36 was also converted into 37 in 70% yield under the same reaction conditions. The stereochemistry of 37 is reminiscent of the natural product tetrodecamycin 6 [7,15].

Epoxide hydration of 25b and formation of the lactone 37.
The bicyclic epoxide 29b, obtained from a DMDO-mediated epoxidation of compound 20, appeared to be unstable and was therefore directly reduced to the corresponding alcohol 38 using the Luche conditions [16], thus furnishing another functional triad toward the oxygenated bridges of harringtonolide 5 (Scheme 8) [9]. However, the stereochemistry of this substrate precluded any direct cascade cyclization starting from the hydroxyl group. Several Lewis and Brønsted acids were used to promote epoxide opening and cyclization events. Especially, initiation of the cyclization from the ethyl ester by activation of the epoxide with a halogenated Lewis acid was attempted, but unsuccessfully. In particular, titanium tetrachloride led to the oxetane 40, probably arising from coordination of the free hydroxyl group and generation of an allylic cation quenched by a transient chlorohydrine (39). Protection of the hydroxyl group of 38 (TBS) stopped the reaction at the chlorohydrine stage.
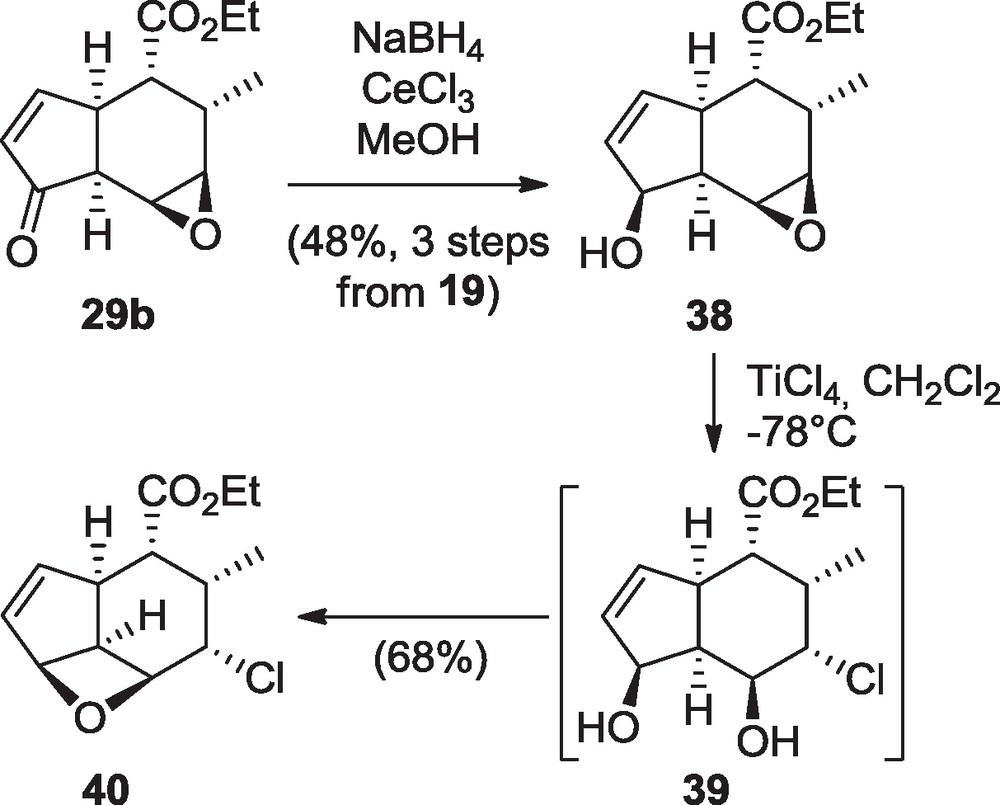
Reactivity of the functional triad 38 in the presence of TiCl4.
With substrate 38, the best Brønsted acid conditions involved potassium hydrogenosulfate in combination with ytterbium triflate, which enhanced the rate of the reaction. Brønsted acid assisted Lewis acids (BLA) [17] have been used for Diels-Alder reactions, and more specifically ytterbium salts have been used in combination with a carboxylic acid for ene-like reactions [18] or for allylation of aldehydes [19]. The reaction would involve a dual activation complex for pinching the Lewis base (Scheme 9 represents our BLA system).
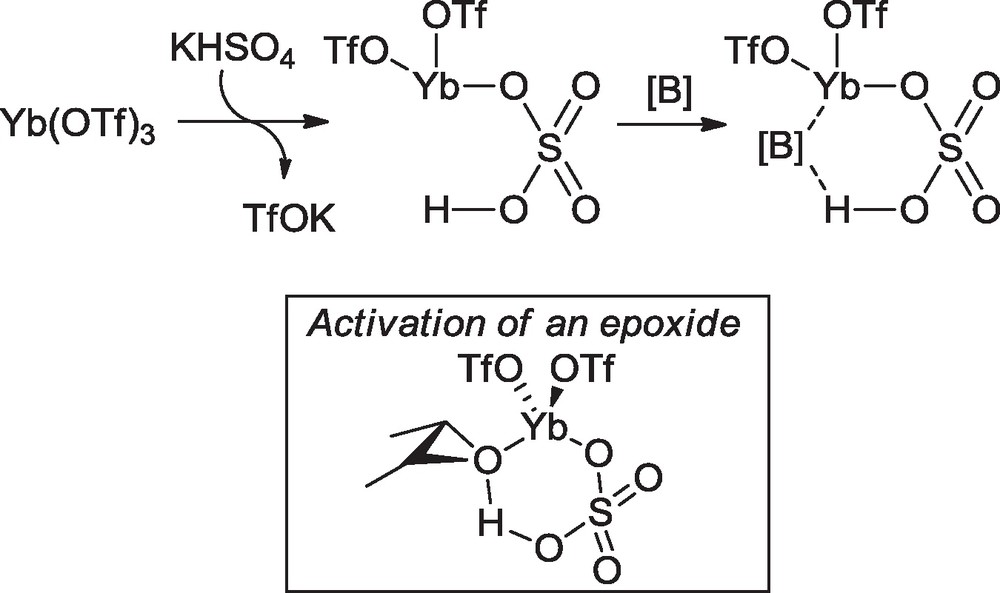
Mechanism of a dual Brønsted-Lewis acid-catalysis (Yb(OTf)3–KHSO4 in our case): the Lewis base [B] could be an epoxide (box).
We found that this combination of acids (Yb(OTf)3, KHSO4) in THF at 50 °C was able to promote the regioselective epoxide opening of 38 and at the same time in situ closing of the lactone and cyclic ether bridges, to get the polycyclic system 41 in 39% yield in a one-pot process (Scheme 10). Occasionally, the intermediate triol 42 could be isolated, especially when the reaction was performed in water in the presence of Yb(OTf)3 supported on silica (56% yield) [20]. No other regioisomer of epoxide hydration could be identified, showing that this reaction is highly regioselective, either due to the pseudo-axial methyl substituent having a congesting effect of the nearest carbon C-6 (hydration occurs on C-7) or to discrimination of both epoxide carbones by the BLA complex. The reaction gratifyingly offers access to an important structure part of the natural product harringtonolide (5), confirmed by 2D NMR analysis and X-ray crystallography [9].

Action of the “BLA” Yb(OTf)3–KHSO4 on substrate 38: efficient access to the oxygenated polycyclic structure of harringtonolide 5.
5 Conclusion
We have constructed various hydroxy-epoxy-ester triads for cascade reactions toward polycyclic oxygen-bridged cage structures. Such structures were obtained under acid-catalyzed conditions, the outcome of which mainly depends on the epoxide configuration and on the type of acid catalyst. If applicable, the reaction always followed the Baldwin rules, while when the reaction was blocked by stereochemical constraints, opening (hydration, chlorination) of the epoxide was first needed. Various and complex polycyclic structures were thus obtained during this work, culminating with the oxygen-bridged cage structure of harringtonolide (5) obtained from a reaction cascade promoted by a dual Brønsted-Lewis acid complex.
Acknowledgements
The Agence nationale de la recherche (ANR) is gratefully acknowledged for funding this project and the PhD grant for HA (grant number ANR-09-JCJC-0085-01). The Ministère de l’Éducation Nationale et de la Recherche is acknowledged for providing a PhD scholarship to LE. The Centre national de la recherche and the Muséum national d’Histoire naturelle are acknowledged for daily supporting this work.
2 This lettering of cycles differs from our original publication [9] and follows the putative biosynthetic nomenclature as described in [6b].
3 Unfortunately, due to limited quantities of the substrates, not all the epoxides could be engaged in the reactions.