

1 Introduction
Highly selective and atom-economic reactions can be reached using catalysis but challenges remain for some reactions like the synthesis of amines through hydroamination reactions [1]. The aza-Michael reaction involving the reaction of activated alkenes and amines can be considered as a hydroamination reaction. It was largely applied in organic synthesis with the use of various metal or organic catalysts [2]. Surprisingly, the uncatalysed “background” aza-Michael reaction has only been scarcely reported [3,4] and, to the best of our knowledge, no comprehensive study has been carried out so far. Such data could be of interest for highly selective aza-Michael reactions through a rational choice of reagents and experimental conditions. Herein, we would like to report on advantages and limits of uncatalysed aza-Michael reactions. The reactivity scope will be exposed leading to original mono- and di-hydroamination products.
2 Results and discussion
As a start, several weak amine nucleophiles were allowed to react with 10 equivalents of methylvinylketone 1 (Table 1). The use of an excess of alkene to react with an amine is known to increase drastically the conversions; indeed, such a reactivity enhancement was already observed for several catalysed hydroamination reactions of unactivated alkenes [1c,d,g].
Amine reactivity towards methylvinylketone 1.
| Entry | Equivalents of alkene 1 | Amine | Conversion (%)a |
| 1 | 10 | 0 | |
| 2 | 10 | 0 | |
| 3 | 10 | 0 | |
| 4 | 10 | 0 | |
| 5 | 10 | 77b | |
| 6 | 10 | 100 (79 + 21)c | |
| 7 | 2 | 76 (60 + 16)c | |
| 8 | 1 | 71 (64 + 7)c |
a Conversions measured by 1H NMR.
b Performed in toluene at 100 °C for 40 h.
c Yields of compounds 3f1 (branched) + 3f2 (linear).
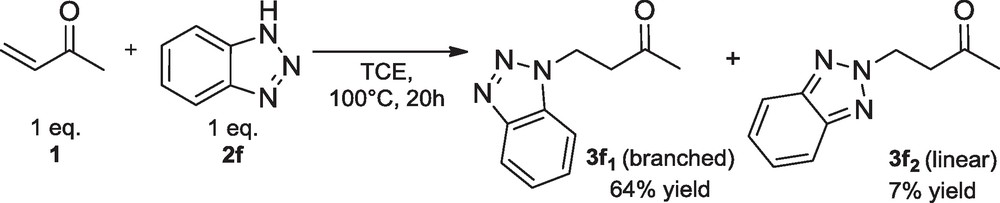
In order to check the reactivity issues at a sufficient level of activation, reactions were performed at 100 °C in polar or apolar solvents, respectively tetrachloroethane (TCE) or toluene. Reaction of 1 proceeded with imine 2e to lead to product 3e most likely because of higher amine nucleophilicity (entry 5). The use of toluene instead of TCE proved to increase drastically the yield of 3e, acid traces in TCE decomposing imine 2e. By comparing reactivity and pKa values in DMSO of the involved amine reagents, imine 2e (pKa 31.0) was found to be the strongest nucleophile of that series as compared to benzamide 2a (pKa 23.3), oxazolidone 2b (pKa 20.8) or the amines 2c-d,f. By allowing a 71% yield of products 3f1 and 3f2, benzotriazole 2f (pKa 11.9) first appeared to be an exception regarding the poor nucleophilicity of 2f (Table 1, entry 8 and Scheme 1). It was worth noting the reaction was quantitative as respect to the amine by using 10 equivalents of alkene 1 (entry 6). Moreover, the observed rearrangement of branched product 3f1 into linear product 3f2 was already reported in other syntheses [5].
However, Wang et al. recently described catalyst-free aza-Michael reactions of azoles with β,γ–unsaturated-α-keto esters [3e]. The hydrogen bonding between the carbonyl oxygen of the activated alkene and the NH moiety of the N-heterocyclic nucleophile was proposed to form the key reaction intermediate. Then, the fully conjugate nucleophile addition afforded an enol intermediate and later the final addition product after a retro-enolization. Hence, as a general trend, the catalyst-free reactions of amines with activated alkenes appeared to depend on the strength of the nucleophile. Nevertheless, in spite their poor pKas, N-heterocyclic amines could react with different activated alkenes affording valuable intermediates. Such results tended to demonstrate the hydrogen-bonding interactions between activated alkenes and such poly-nitrogen aromatic cycles may control these concerted or fully conjugate aza-Michael additions.
Next, we studied the reactivity of acrylate derivatives 4a,b with primary and secondary amines 2f-s at 100 °C and 30 °C in toluene. The last was preferred to TCE as far as basic amines proved to be more sensitive to acidic residues of chlorinated solvents (Table 2).
Primary and secondary amine reactivity towards acrylate derivatives 4a,b.
| Entry | Amine | Alkene (eq.) | Yield (%) 5(f-t)(a-b) at 100 °Ca | Yield (%) 6(n-t)(a-b) at 100 °Ca | Yield (%) 5(f-t)(a-b) at 30 °Ca,b |
| 1c | 4a (10 eq.) | 15 (6 + 9)f (5fa1 + 5fa2) | 0 | < 5 (5fa)g | |
| 2 | 4b (10 eq.) | < 5 (5fb1)g | 0 | 0 | |
| 3c,d | 4a (10 eq.) | 70 (5ia) | 0 | < 5 (5ia)g | |
| 4 | 4b (10 eq.) | 77 (5ib) | 0 | – | |
| 5e | 2j (nBu)2NH | 4a (10 eq.) | 90 (5ja) | 0 | 15 (5ja) |
| 6 | 2k MeBnNH | 4a (10 eq.) | 96 (5ka) | 0 | 55 (5ka) |
| 7 | 2l MePhNH | 4a (10 eq.) | 0 | 0 | 0 |
| 8 | 2m (iPr)2NH | 4a (10 eq.) | 0 | 0 | 0 |
| 9 | 2n iPrNH2 | 4a (10 eq.) | 37 (5na)h | 38 (6na) | 8 (5na) |
| 10 | 2o CyNH2 | 4a (10 eq.) | 100 (5oa) | 0 | 23 (5oa) |
| 11 | 2p BnNH2 | 4a (10 eq.) | 58 (5pa) | 42 (6pa) | 38 (5pa) |
| 12 | 2p BnNH2 | 4b (10 eq.) | 95 (5pb) | 0 | – |
| 13 | 2q PhNH2 | 4a (10 eq.) | 0 | 0 | – |
| 14 | 2r nBuNH2 | 4a (10 eq.) | 0 | 90 (6ra) | 44 (5ra) |
| 15 | 2s tBuNH2 | 4a (10 eq.) | 32 (5sa) | 0 | 0 |
| 16 | 2t MeNH2 | 4a (10 eq.) | 0 | 42 (6ta)i | 0 |
| 17 | 2p BnNH2 | 4a (5 eq.) | 84 (5pa) | 12 (6pa) | 25 (5pa)g |
| 18 | 2p BnNH2 | 4a (2 eq.) | 83 (5pa) | 11 (6pa) | – |
| 19 | 2p BnNH2 | 4a (1 eq.) | 99 (5pa) | 0 | – |
a Isolated yield; all reactions performed with 0.5 mmol of amine.
b 20 h reactions.
c TCE as solvent.
d 23 h reaction.
e 26 h reaction.
f Compounds 5fa1 (branched) and 5fa2 (linear).
g Measured by 1H NMR.
h 40 h reactions.
i Same result at 30 °C.
With the exception of amine 2f which afforded poor yields of compounds 5fa1, 5fa2 and 5fb1 (entries 1–2), the reactions proceeded quite well for secondary amines 2i-k at 100 °C affording mono-hydroamination products 5ia, 5ib, 5ja and 5ka in good yields (entries 3–6). For these last products, yields were quite low at 30 °C except for reaction with N-methylbenzylamine 2k (entry 6). Regarding N-methylaniline 2l and diisopropylamine 2m (entries 7,8), no conversion was obtained. On the whole, these results confirmed that the less the nitrogen atom was hindered, the more the hydroamination reaction was enabled. The reactivity of primary amines 2n-t with acrylates 4a,b was then investigated in detail. At 100 °C, mono-hydroamination 5(n-t)a and di-hydroamination 6(n-t)a products were obtained with average to good yields (entries 9–16). It was worth noting methyl crotonate 4b afforded only mono-hydroamination product 5pb (entry 12) and aniline 2q did not react (entry 13). Strikingly, cyclohexylamine 2o, tertiobutylamine 2s led selectively to mono-hydroamination products 5oa and 5sa (entries 10, 15) whereas butylamine 2r and methylamine 2t afforded only di-hydroamination products 6ra and 6ta (entries 14, 16). Hence, we could state a sterically bulky primary amine reagent would privilege a mono-hydroamination product whereas a less hindered amine would mainly lead to a di-hydroamination product. However, γ-substituted activated alkenes would offer exclusively mono-hydroamination products. Moreover, lowering the temperature to 30 °C induced the selective formation of mono-hydroamination compounds 5na, 5oa, 5pa and 5ra but in low yields (entries 9–11, 14). The use of a large excess of alkene 4a proved to be crucial in order to reach average to good yields of mono-hydroamination product 5pa and di-hydroamination product 6pa (entries 11, 17–19). To the best of our knowledge, such results are rare examples of controlled di-hydroamination reactions [6].
Finally, the reaction of compounds 4a and 2p using 10 mol% of mono-hydroamination compound 5pa as catalyst afforded 5pa in a 17% yield which was lower than the 38% yield of the reaction run without catalyst at 30 °C (Table 2, entry 11). Hence, product 5pa could not perform any autocatalysis for the reaction of reagents 4a and 2p [7]. Moreover, reactions of amine 2p with alkene 4a proved to be sensitive to the nature of the solvent used, neat reaction running also very well (Table 3, entries 1, 6–10). It is worth noting the yields of 5pa were drastically increased by the addition of water to the reaction medium (entries 2, 3). The use of protic additives like hydroquinone and silica did also enhance the yields of 5pa and small amounts of 6pa could be isolated (entries 4, 5) [8]. Such rate enhancement in aza-Michael reactions, though known but rarely quoted, can be described as an external proton transfer activation by protic media [6d,8a].
Solvent and additive effects on the reaction of butylacrylate 4a and benzylamine 2p.
| Entry | Solvent | Additive | Yield (%)a 5pa | Yield (%)a 6pa |
| 1 | Toluene | – | 38 | 0 |
| 2 | Toluene | Undistilled alkene | 59 | 0 |
| 3 | Toluene | H2O (2 eq.) | 65 | 0 |
| 4 | Toluene | hydroquinone (10 mol%) | 90 | 10 |
| 5 | Toluene | Silica (10 mol%) | 87 | Traces |
| 6 | Dioxane | – | 37 | 0 |
| 7 | THF | – | 31 | 0 |
| 8 | Et2O | – | 43 | 0 |
| 9 | DCM | – | 62 | 0 |
| 10 | None | – | 85 | 9 |
a Alkene and solvent were distilled. Amine was not distilled.
In order to clarify the scope of these catalyst-free aza-Michael reactions, we next studied reactions of different activated alkenes with an amine. Considering the raising interest on conjugate addition reactions of nitrogen-centered heterocyclic nucleophiles to electron-deficient olefins [3e,9], we choose benzotriazole 2f which is also a useful synthetic auxiliary (Table 4) [10]. At 100 °C in TCE, reactions of 2f with alkenes 7a-f afforded addition products 8a1-f1 in good yields (Table 4, entries 1–6). Moreover, alkenes 7a, 7c, 7d, 7f afforded rearranged products 8a2, 8c2, 8d2, 8f2 in low yields (entries 1, 3, 4, 6) [5]. At 30 °C in dichloromethane, the uncatalysed reactions using alkenes 7a-c were almost quantitative and there was no evidence for any polymerisation of the alkene reagents at such a reaction temperature according NMR spectra of the corresponding crude mixtures (Table 4, entries 1–3). However, at 30 °C, reactions of benzotriazole 2f with alkenes 7d-f offered lower conversions or none (Table 4, entries 4–6). Finally, no reactivity was observed for ethylcrotonate, acrylonitrile, 2-butenitrile, 3-penten-2-one, ethylcinnamate and phenyltrans-styryl sulphone, independently of the reaction conditions used.
Benzotriazole reactivity towards activated alkenes.
| Entry | Alkene 7a-f | Time (h) | Yield (%) at 100 °Ca | Conversion (%) at 30 °Cb |
| 1 | 20 | 86c (69 + 17) (8a1, 8a2) | 100c (84 + 26) (8a1, 8a2) | |
| 2 | 20 | 82 (8b1) | 94 (8b1) | |
| 3 | 20 | 97c (84 + 13) (8c1, 8c2) | 86 (8c1) | |
| 4 | 20 | 69c (46 + 23) (8d1, 8d2) | < 5 (8d1) | |
| 5 | 65 | 51 (8e1) | 11d (8e1) | |
| 6 | 65 | 100c (83 + 17) (8f1, 8f2) | – |
a Isolated yield.
b Performed in CH2Cl2 for 30 h, conversions by 1H NMR.
c Products 5fa1, 8a1,c1,d1,f1 from addition and 5fa2, 8a2,c2,d2,f2 from rearrangement.
d 20 h reaction.
3 Conclusion
The studied aza-Michael reactions were shown to be performed without the use of any catalyst depending on a combination of different factors. The strength of the nucleophile appeared to be the key parameter to control the reactivity, temperature and steric hindrance of the reagents being the other factors mastering the selectivity of these mono- and di-hydroamination reactions. The last were shown to proceed only when heat and large excesses of activated alkenes were used. This allowed the synthesis of rare products resulting from a double-addition of an amine on two equivalents of an activated alkene. Moreover, the catalyst-free conjugate addition reaction of various electron-deficient olefins and poor nucleophiles like N-heterocyclic amines was demonstrated and afforded valuable intermediates for organic synthesis. Such results tended to demonstrate the hydrogen-bonding interactions between activated alkenes and poly-nitrogen aromatic cycles may control these concerted or fully conjugate aza-Michael additions. Finally, when a catalysed aza-Michael reaction is performed, it may be asked whether a strict catalysed reaction is involved or if any competitive background reaction is taking place.
4 Experimental
4.1 General remarks
All solvents were dried using standard methods. Alkenes were distilled under vacuum over CaH2 and stored over molecular sieves (4 Å). Amine substrates were placed under vacuum during one hour before use, or distilled over CaH2 if they were liquid. All reactions were carried out under a dry nitrogen atmosphere. Analytical thin layer chromatography (TLC) was performed on Merck pre-coated 0.20 mm silica gel Alugram Sil 60 G/UV254 plates. Flash chromatography was carried out with Macherey silica gel 60 M. 1H (300 MHz or 400 MHz), 13C (75 MHz) and 31P (121 MHz) NMR spectra were acquired on Bruker Avance spectrometers. Chemical shifts are reported downfield of Me4Si and coupling constants are expressed in Hz. 1,3,5-trimethoxybenzene and 1,2,4,5-tetrachlorobenzene were used as internal standards when needed. Gas chromatography analyses were done on GC Varian 3900 and 430 using Alltech EconoCap EC-5™ column (30 m, 0.25 mm, 0.25 μm) with program (5 °C/min, 100–230 °C, 36 min) and with tetradecane as internal standard. GC-MS analyses were performed on a Shimadzu QP2010 + using Supelco column SLB™-5ms (30 m, 0.25 mm, 0.25 μm). Infrared spectra were recorded on a ThermoScientific-Nicolet 6700 spectrometer; the samples being prepared with KBr powder. HRMS-ESI analyses were performed at CUMA-University Lille Nord de France. Elemental analyses were performed on a Elementar Vario Micro Cube apparatus at UCCS, University Lille Nord de France. See supporting information for other details.
4.2 General procedures
4.2.1 General procedure for amine reactivity screening with methylvinylketone
Amine substrate (0.5 mmol) (if solid) was added in a Schlenk flask and dried under vacuum for 30 minutes. Next, dry 1,1′,2,2′-tetrachloroethane or toluene (1 mL), methylvinylketone (5.0 mmol, 0.4 mL), and amine substrate (0.5 mmol) (if liquid) were added. After stirring at 100 °C for the corresponding time, the mixture was concentrated under vacuum, and the resulting product was isolated by flash chromatography.
4.2.2 General procedure for amine substrate screening
Amine substrate (0.5 mmol) (if solid) was added in a Schlenk flask and dried during 30 minutes under vacuum. Then, dry toluene (1 mL), butylacrylate (5.0 mmol, 0.7 mL), and freshly distilled amine substrate (0.5 mmol) (if liquid) were added under a nitrogen atmosphere. After stirring at 100 °C for the corresponding time, the mixture was concentrated under vacuum, and the resulting product was isolated by flash chromatography.
4.2.3 General procedure for activated alkene screening
Benzotriazole (0.5 mmol, 59.6 mg) and activated alkene (5.0 mmol) (if solid) were added in a Schlenk flask and dried during 30 min under vacuum. Then, dry 1,1′,2,2′-tetrachloroethane (1 mL), and activated alkene (5.0 mmol) (if liquid) were added under a nitrogen atmosphere. After stirring at 100 °C at the corresponding time, the mixture was concentrated under vacuum, and the corresponding product was isolated by flash chromatography.
4.3 Physical and spectra data for 5ja, 5ka, 6na, 6pa, 6ra, 6ta, 8e1, 8f1, 8f2
4.3.1 Butyl-3-(dibutylamino)propanoate 5ja
Isolated after flash chromatography with petroleum ether/NEt3 (9:1), as a colourless oil (from 0.5 mmol, 0.45 mmol-116 mg, 90% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.85–0.95 (q*, 9H), 1.20–1.45 (m, 10H), 1.58 (quin, J(H,H) = 7.2 Hz, 2H), 2.30–2.45 (m, 6H), 2.75 (t, J(H,H) = 7.5 Hz, 2H), 4.05 (t, J(H,H) = 6.7 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ = 13.7 (CH3), 14.1 (2 CH3), 19.1 (CH2), 20.6 (2 CH2), 29.3 (2 CH2), 30.7 (CH2), 32.4 (CH2), 49.4 (CH2), 53.6 (2 CH2), 64.2 (CH2), 173.1 (C). IR (KBr): ν∼ = 2959, 2933, 2873, 1717, 1202 cm−1. HMRS (ESI) m/z calcd for C15H32NO2: 258.2428 [MH+], found: 258.2423.
4.3.2 Butyl-3-(benzyl(methyl)amino)propanoate 5ka
Isolated after flash chromatography with petroleum ether/ethylacetate/NEt3 (9:1:1) (Rf = 0.75) as a colourless oil (from 0.5 mmol, 0.48 mmol-120 mg, 96% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.95 (t, J(H,H) = 7.4 Hz, 3H), 1.39 (sex, J(H,H) = 7.4 Hz, 2H), 1.63 (quin, J(H,H) = 7.1 Hz, 2H), 2.22 (s, 3H), 2.54 (t, J(H,H) = 7.2 Hz, 2H), 2.77 (t, J(H,H) = 7.2 Hz, 2H), 3.53 (s, 2H), 4.10 (t, J(H,H) = 6.7 Hz, 2H), 7.20–7.40 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ = 13.8 (CH3), 19.2 (CH2), 30.7 (CH2), 33.0 (CH2), 41.8 (CH3), 52.9 (CH2), 62.1 (CH2), 64.3 (CH2), 127.0 (CH), 128.2 (2 CH), 129.0 (2 CH), 138.9 (C), 172.7 (C). IR (KBr): ν∼ = 2954, 2841, 2791, 1734, 1124 cm−1. HMRS (ESI) m/z calcd for C15H24NO2: 250.1802 [MH+], found: 250.1798.
4.3.3 Dibutyl-3,3′-(isopropylazanediyl)dipropanoate 6na
Isolated after flash chromatography with petroleum ether/NEt3 (9:1) as a colourless oil (from 0.50 mmol, 0.19 mmol-60 mg, 38% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.85–1.00 (m, 12H), 1.34 (sex, J(H,H) = 7.5 Hz, 4H), 1.57 (quin, J(H,H) = 6.9 Hz, 4H), 2.38 (t, J(H,H) = 7.2 Hz, 4H), 2.68 (t, J(H,H) = 7.2 Hz, 4H), 2.85 (sep, J(H,H) = 6.6 Hz, 1H), 4.03 (t, J(H,H) = 6.6 Hz, 4H). 13C NMR (CDCl3, 75 MHz): δ = 13.8 (2 CH3), 18.4 (2 CH3), 19.2 (2 CH2), 30.8 (2 CH2), 34.8 (2 CH2), 45.8 (2 CH2), 50.7 (CH), 64.2 (2 CH2), 173.0 (2 C). IR (KBr): ν∼ = 2979, 1733, 1137 cm−1. HMRS (ESI) m/z calcd for C17H34NO4: 316.24824 [MH+], found: 316.24873.
4.3.4 Dibutyl-3,3′-(benzylazanediyl)dipropanoate 6pa
Isolated after flash chromatography with petroleum ether/ethylacetate/NEt3 (8:2:1), Rf = 0.7, as a colourless oil (from 0.50 mmol, 0.21 mmol-76 mg, 42% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.85 (t, J(H,H) = 7.3 Hz, 6H), 1.28 (sex, J(H,H) = 7.3 Hz, 4H), 1.49 (quin, J(H,H) = 6.9 Hz, 4H), 2.38 (t, J(H,H) = 7.2 Hz, 4H), 2.73 (t, J(H,H) = 7.2 Hz, 4H), 3.52 (s, 2H), 3.98 (t, J(H,H) = 6.7 Hz, 4H), 7.15–7.25 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ = 13.7 (2 CH3), 19.1 (2 CH2), 30.6 (2 CH2), 32.7 (2 CH2), 49.2 (2 CH2), 58.2 (CH2), 64.3 (2 CH2), 127.0 (CH), 128.2 (2 CH), 128.7 (2 CH), 139.1 (C), 172.7 (2 C). IR (KBr): ν∼ = 2959, 2873, 1734, 1176 cm−1. HMRS (ESI) m/z calcd for C21H34NO4: 364.2482 [MH+], found: 364.2475.
4.3.5 Dibutyl-3,3′-(butylazanediyl)dipropanoate 6ra
Isolated after flash chromatography with petroleum ether: NEt3 (9:1) as a colourless oil (from 0.5 mmol, 0.45 mmol-148 mg, 90% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.80–0.95 (m, 9H), 1.15–1.40 (m, 8H), 1.55 (quin, J(H,H) = 7.5 Hz, 4H), 2.35 (q, J(H,H) = 7.5 Hz, 6H), 2.71 (t, J(H,H) = 7.3 Hz, 4H), 4.01 (t, J(H,H) = 6.7 Hz, 4H). 13C NMR (CDCl3, 75 MHz): δ = 13.7 (2 CH3), 14.0 (CH3), 19.1 (2 CH2), 20.4 (CH2), 29.3 (CH2), 30.6 (2 CH2), 32.6 (2 CH2), 49.2 (2 CH2), 53.4 (CH2), 64.2 (2 CH2), 172.8 (2 C). IR (KBr): ν∼ = 2960, 2934, 2873, 1734, 1189 cm−1. HMRS (ESI) m/z calcd for C18H36NO4: 330.2639 [MH+], found: 330.2632.
4.3.6 Dibutyl-3,3′-(methylazanediyl)dipropanoate 6ta
Isolated after flash chromatography with petroleum ether/NEt3 (9:1) as a colourless oil (from 0.5 mmol, 0.2 mmol-60 mg, 42% yield). 1H NMR (CDCl3, 300 MHz): δ = 0.88 (t, J(H,H) = 7.2 Hz, 6H), 1.34 (hex, J(H,H) = 7.2 Hz, 4H), 1.55 (quin, J(H,H) = 6.9 Hz, 4H), 2.20 (s, 3H), 2.42 (t, J(H,H) = 7.2 Hz, 4H), 2.66 (t, J(H,H) = 7.2 Hz, 4H), 4.03 (t, J(H,H) = 6.6 Hz, 4H). 13C NMR (CDCl3, 75 MHz): δ = 12.8 (2 CH3), 18.2 (2 CH2), 29.7 (2 CH2), 31.7 (2 CH2), 40.8 (CH3), 51.6 (CH2), 63.3 (2 CH2), 171.7 (CO). IR (KBr): ν∼ = 2960, 2930, 2850, 1738, 1180 cm−1.
4.3.7 1-(2-(phenylsulfonyl)ethyl)-1H-benzo[d][1,2,3]triazole 8e1
Isolated after flash chromatography with petroleum ether/ethylacetate (1:1), Rf = 0.3, as a white solid (from 0.50 mmol, 0.25 mmol-72 mg, 51% yield). M.p. = 97–99 °C. 1H NMR (CDCl3, 300 MHz): δ = 3.89 (t, J(H,H) = 6.9 Hz, 2H), 5.01 (t, J(H,H) = 6.9 Hz, 2H), 7.30–7.45 (m, 3H), 7.45–7.60 (m, 3H), 7.71 (d, J(H,H) = 7.8 Hz, 2H), 7.94 (d, J(H,H) = 8.3 Hz, 1H). 13C NMR (CDCl3, 75 MHz): δ = 41.5 (CH2), 54.9 (CH2), 109.13 (CH), 120.1 (CH), 124.3 (CH), 127.6 (2 CH), 127.9 (CH), 129.3 (2 CH), 132.9 (C), 134.1 (CH), 138.2 (C), 145.7 (C). IR (KBr): ν∼ = 2989, 1308, 1140 cm−1. HMRS (ESI) m/z calcd for C14H14N3O2S: 288.08012 [MH+], found: 288.08004.
4.3.8 3-(3-(1H-benzo[d][1,2,3]triazol-1-yl)butanoyl)oxazolidin-2-one 8f1
Isolated after flash chromatography with petroleum ether/ethylacetate (8:2), Rf = 0.3, as a white solid (from 0.50 mmol, 0.42 mmol-114 mg, 83% yield). M.p. = 123–125 °C. 1H NMR (CDCl3, 300 MHz): δ = 1.70 (d, J(H,H) = 6.9 Hz, 3H), 3.47 (dd, J(H,H) = 4.6 Hz and J(H,H) = 18.2 Hz, 1H), 3.70-3.95 (m, 2H), 4.08 (dd, J(H,H) = 9.0 Hz and J(H,H) = 18.2 Hz, 1H), 4.30 (sex, J(H,H) = 8.4 Hz, 2H), 5.35–5.50 (m, 1H), 7.31 (t, J(H,H) = 7.5 Hz, 1H), 7.45 (t, J(H,H) = 7.5 Hz, 1H), 7.62 (d, J(H,H) = 8.4 Hz, 1H), 7.98 (d, J(H,H) = 8.4 Hz, 1H). 13C NMR (CDCl3, 75 MHz): δ = 21.2 (CH3), 41.4 (CH2), 42.3 (CH2), 50.6 (CH), 62.3 (CH2), 109.9 (CH), 119.8 (CH), 124.0 (CH), 127.2 (CH), 132.7 (C), 145.9 (C), 153.5 (C), 169.8 (C). IR (KBr): ν∼ = 2985, 2928, 1777, 1698, 1221, 1120 cm−1. HMRS (ESI) m/z calcd for C13H15N4O3: 275.11387 [MH+], found: 275.1139.
4.3.9 3-(3-(2H-benzo[d][1,2,3]triazol-2-yl)butanoyl)oxazolidin-2-one 8f2
Isolated after flash chromatography with petroleum ether/ethylacetate (8:2), Rf = 0.5, as a white solid (from 0.500 mmol, 0.085 mmol-23 mg, 17% yield). M.p. = 82–84 °C. 1H NMR (CDCl3, 300 MHz): δ = 1.75 (d, J = 6.8 Hz, 3H), 3.52 (dd, J = 4.7 Hz and J(H,H) = 18 Hz, 1H), 3.85–4.00 (m, 2H), 4.04 (dd, J(H,H) = 8.8 Hz and J(H,H) = 18 Hz, 1H), 4.30–4.45 (m, 2H), 5.50–5.65 (m, 1H), 7.34 (dd, J(H,H) = 3.0 Hz and J(H,H) = 6.5 Hz, 2H), 7.83 (dd, J(H,H) = 3.0 Hz and J(H,H) = 6.5 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ = 21.6 (CH3), 41.5 (CH2), 42.3 (CH2), 58.6 (CH), 62.3 (CH2), 118.1 (2 CH), 126.2 (2 CH), 144.0 (2 C), 153.5 (C), 169.7 (C). IR (KBr): ν∼ = 2958, 2927, 1780, 1703, 1275, 1116 cm−1. HMRS (ESI) m/z calcd for C13H15N4O3: 275.11387 [MH+], found: 275.1135.
Acknowledgements
This work is first financed by the French National Research Agency with project ANR-09-BLAN-0032-02 (with a PhD fellowship to F.M.). Support from CNRS is warmly acknowledged. Partial funding from Région Nord-Pas de Calais with “Projet Prim: État-Région” and with “Fonds européen de développement régional (FEDER)” are also appreciated. Mrs C. Méliet (UCCS) is thanked for elemental analyses. Ms C. Delabre (UCCS) is thanked for GC-MS analyses.