

1 Introduction
Since its discovery in 1985 [1], the hollow spherical form of carbon allotropes has attracted increasing attention due to its potential applications. This allotrope was named fullerene. C60 is the most plentiful and smallest stable fullerene with Ih symmetry. The construction principle of the fullerenes is an outcome of the isolated pentagon rule (IPR) [2] and Euler theorem. Based on these principles, for the closure of each spherical network with n hexagons, 12 pentagons are required (n ≠ 1) and none of the pentagons are in contact with each other. Compared to graphene sheets and carbon nanotubes (CNTs), the structures of these three-dimensional systems are wonderful and attractive. Spherical shape of fullerenes is due to the pentagons which are absent in graphene and CNT. Due to the spherical shape of C60, the conjugate π bond should be less effective on the wall of a C60 and there is hybridization between the π and σ orbitals. Therefore, conjugate π bonding among the carbon atoms is limited and this phenomenon causes high reactivity of C60. On-ball doped (dopant is included in the fullerene shell) fullerenes in which one or more of carbon atoms that form the cage structure are replaced by heteroatoms are called heterofullerenes [2–4]. BN is a binary compound made of Group III and Group V elements in the periodic table that is much closer to the carbon system. BN materials are the best candidates to replace carbon because they are isoelectronic with their all-carbon analogues. Therefore, the inclusion of nitrogen and boron dopants for the formation of heterofullerenes with the general formula of C60-2nBnNn is a promising way to modify the physical and chemical properties of fullerenes [3–5]. Existence of atoms with different electronegativities indicates the high ionicity of the heterofullerenes. The chemical functionalization of nano structures is an effective way to enhance their solubility and change their electronic properties [6]. Moreover, the results of these studies are very important on account of application of these materials for sensing and/or filtering of many hazardous molecules such as CO, HCOH, NH3, NOx, etc. Over the past decade, functionalization of various nanotubes has been extensively studied, while the functionalization of heterofullerenes has been less considered. As one of the early pioneers of theoretical study of functionalized heterofullerenes, the author has studied the chemical functionalization of heterofullerenes with NH3 and NO2 [7–10].
Nitrogen dioxide (NO2) is one of a group of highly reactive oxidant and corrosive gasses known as nitrogen oxides (NOx). This toxic gas is mainly emitted from the motor vehicle exhaust, burning of fossil fuels, petrol and metal refining, power plants, gas stoves, etc. [11–13]. Nitrogen dioxide is an important air pollutant because it contributes to the formation of ground-level ozone, a major component of photochemical smog, which can have significant impacts on human health. NO2 is linked with a number of adverse effects on the respiratory system and is a precursor to nitrates, which contribute to increased respirable particle levels in the atmosphere [14,15]. As a radical, the reaction with NO2 [16] is an interesting test of the reactivity of heterofullerenes. According to my knowledge, no prior theoretical investigations have been reported on the covalent/electrostatic nature of addend-heterofullerene interactions. The present work is a DFT study of functionalization of C30B15N15 with NO2 on the different adsorption sites, with a careful examination of a number of configurations. To get more information about the nature of the interaction, energy densities at the bond critical points (BCP) from Bader's [17] quantum theory of atoms in molecules (QTAIM) [18,19], and natural electron configuration of adsorbing atom using natural bond orbital analysis (NBO) [20] were calculated.
2 Model system and computational details
The initial geometry of C60 was adopted for construction of C30B15N15 as model adsorbent by replacing 15 carbon atoms by nitrogen atoms and another 15 carbon atoms by boron atoms (Fig. 1). Geometry optimizations and calculation of their electronic wave functions were performed at the 6-31G* basis set with B3LYP functional [21,22], using Gaussian 03 software package [23]. This model chemistry for adsorption study in pi-extended systems is economical and accurate with respect to other model chemistries and used in many articles [7–10,24–26]. The AIM computations were carried out using the AIM 2000 program [18,19]. An NBO calculation was performed with the use of the NBO program version 5.0 [20] by the B3LYP method and 6-31G* standard basis set in the optimized structures in order to evaluate the natural electron configuration of adsorbing atom and electron charge transfer between bound gas molecule and C30B15N15.

Schlegel diagram showing the C60 core C-atoms, and the dopant atoms of nitrogen and boron.
3 Results and discussion
For a careful investigation of NO2 adsorption on the exterior surface of a C30B15N15 heterofullerene, different adsorption positions and orientation of the triangular shaped NO2 relative to the C30B15N15 surface must be taken into consideration. Three different adsorption sites for adsorption of NO2 are available, adsorbed gas nearest to a C atom, N atom and B atom. Full geometrical optimization of NO2-C30B15N15 complexes with different orientation, including individual N or O atoms of adsorbed gas or both of them close to the surface, at several adsorption sites show that in the most stable configurations, the adsorbed gas is close to a B atom of C30B15N15. Hence, the same labelling and numbering of C60 as the Schlegel diagram was used to show the three-adsorption sites (Fig. 1). In this figure, adsorption sites pointed out in bold. Other boron sites could be considered but they are equivalent to those three, in such a manner that boron atoms labelled 34, 35, 36, 38 and 39 are in equivalent positions as B.37 (site I) and those at 53, 55, 56, 59 and 60 are equivalent to B.54 (site II). At last, boron atoms of sites 52 and 57 are equivalent to B.58 (site III). Equivalent sites indicate similarity in position symmetry and binding to neighbouring atoms with those three considered sites. On the other hand, there are three possible orientations for adsorption of NO2 on the exterior of a C30B15N15, as schematically illustrated in Figs. 2–4. In the nitro configuration (A), the NO2 molecule is bonded to the surface with the nitrogen end. Bonding with one oxygen end produces the trans-nitrite configuration (B), while bonding with both oxygen ends produces the cis-nitrite configuration (C). For assurance that optimized NO2-C30B15N15 complexes are in global minimum, rotation of adsorbed molecule around the C2 axis was performed, then complexes in minimum state reoptimized at the spin-unrestricted B3LYP/6-31G* level of theory. To evaluate the interaction between a NO2 molecule with different orientations and those three sites of C30B15N15 heterofullerene, binding energies (Eb) computed, according to following:
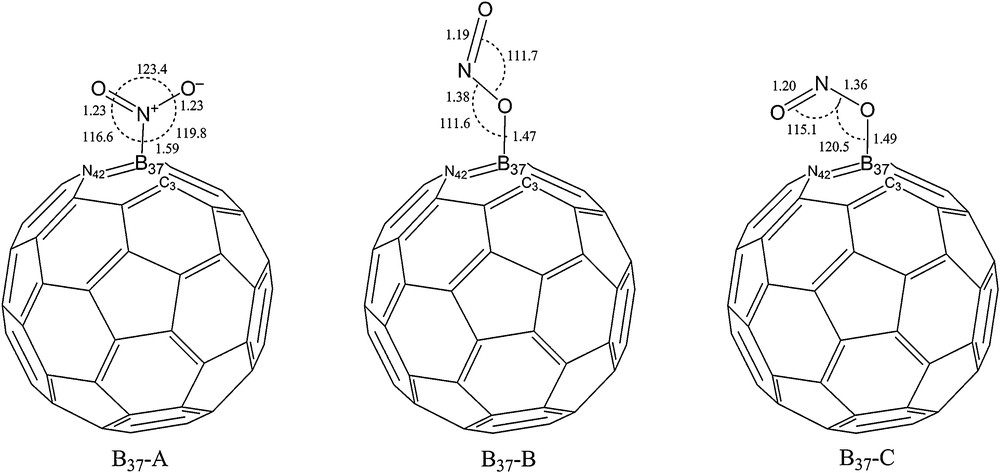
Adsorption of NO2 on the exterior surface of C30B15N15 heterofullerene through B37. The distances shown are in angstroms.

Adsorption of NO2 on the exterior surface of C30B15N15 heterofullerene through B54. The distances shown are in angstroms.
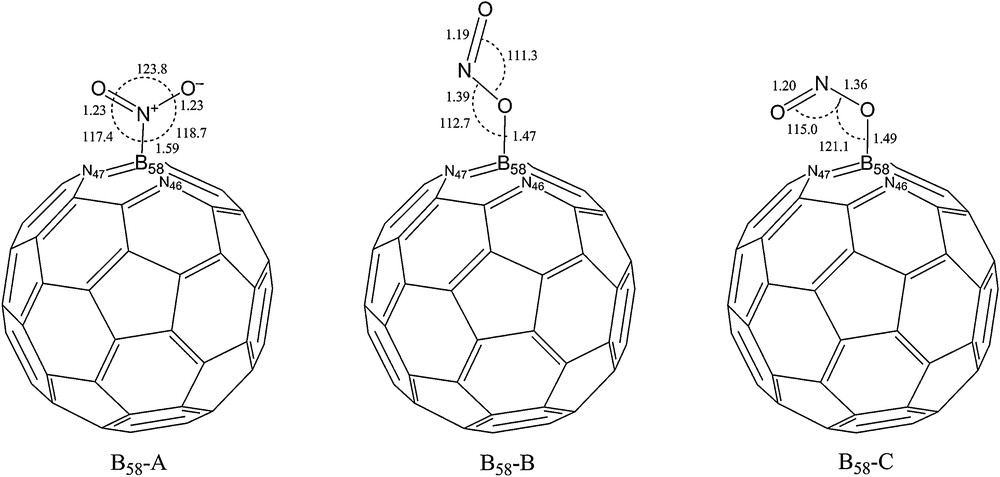
Adsorption of NO2 on the exterior surface of C30B15N15 heterofullerene through B58. The distances shown are in angstroms.
Calculated structural parameters (the distances are in angstroms).
| Model (Configuration) | B.37–C.3 | B.37–N.40 | B.37–N.42 | B.54–C.4 | B.54–N.44 | B.54–N.45 | B.58–C.14 | B.58–N.46 | B.58–N.47 |
| Bare model | 1.54 | 1.43 | 1.47 | 1.55 | 1.44 | 1.47 | 1.55 | 1.45 | 1.46 |
| B37-A | 1.60 | 1.53 | 1.57 | ||||||
| B37-B | 1.60 | 1.55 | 1.59 | ||||||
| B37-C | 1.60 | 1.54 | 1.59 | ||||||
| B54-A | 1.63 | 1.53 | 1.55 | ||||||
| B54-B | 1.64 | 1.55 | 1.57 | ||||||
| B54-C | 1.64 | 1.55 | 1.57 | ||||||
| B58-A | 1.61 | 1.54 | 1.56 | ||||||
| B58-B | 1.63 | 1.56 | 1.59 | ||||||
| B58-C | 1.63 | 1.56 | 1.59 |
Calculated total electronic energies, adsorption energies and charge transfer for adsorption of NO2 on the exterior surface of C30B15N15 heterofullerene.a
| Model (Configuration) | Total Electronic Energy (a.u.) | Eb (kcal/mol)c | QT (el)b |
| Bare Model | –2338.1525321 | ||
| B37-A | –2543.2828574 | –36.47 | –0.449 |
| B37-B | –2543.2947684 | –43.94 | –0.508 |
| B37-C | –2543.2954393 | –44.36 | –0.485 |
| B54-A | –2543. 2879756 | –39.68 | –0.445 |
| B54-B | –2543. 3000198 | –47.24 | –0.499 |
| B54-C | –2543. 2998987 | –47.16 | –0.489 |
| B58-A | –2543. 2891551 | –40.43 | –0.439 |
| B58-B | –2543. 3003500 | –47.45 | –0.505 |
| B58-C | –2543. 3003660 | –47.46 | –0.486 |
a Eele (NO2) = –205.0722062 Hartree.
b Charge transfer from C30B15N15 to NO2 molecule.
c The binding energy between the NO2 molecule and C30B15N15.
Calculated natural electron configuration of adsorbing B atom.
| Model (Configuration) | Natural Electron Configuration | ||
| B37 | B54 | B58 | |
| Bare Model | [core] 2S0.52 2p1.44 ≈ sp2.77 | [core] 2S0.52 2p1.45 ≈ sp2.79 | [core] 2S0.52 2p1.42 ≈ sp2.73 |
| A | [core] 2S0.51 2p1.61 ≈ sp3.15 | [core] 2S0.51 2p1.61 ≈ sp3.15 | [core] 2S0.52 2p1.60 ≈ sp3.07 |
| B | [core] 2S0.49 2p1.50 ≈ sp3.06 | [core] 2S0.49 2p1.48 ≈ sp3.02 | [core] 2S0.50 2p1.46 ≈ sp2.92 |
| C | [core] 2S0.49 2p1.52 ≈ sp3.10 | [core] 2S0.50 2p1.50 ≈ sp3.00 | [core] 2S0.51 2p1.49 ≈ sp2.92 |
For negative H(r), the local potential energy density will dominate and an accumulation of electronic charge in the internuclear region will be stabilizing. This case is a covalent bond. For zero or positive H(r), there will be closed-shell or electrostatic interactions [28]. The values of ▿2ρ(r), G(r), V(r) and H(r) are presented in Table 4. For nine studied NO2-C30B15N15 complexes, the Laplacian of total electronic densities at BCPs are positive and reveals that electronic charges are depleted in the interatomic path, which is characteristic of closed-shell interactions, but according to above theoretical background, this discussion is not valid because in the all studied cases 2G(r) > |V(r)| > G(r). Negative values of H(r) emphasizing that interaction of NO2 and C30B15N15 in all studied gas-heterofullerene complexes are covalent in nature.
Topological parameters calculated at the BCP between NO2 and C30B15N15 on the B3LYP/6-31G* wave functions.
| Model (Configuration) | ▿2ρ(r) | G(r) eV | V(r) eV | H(r) eV |
| B37-A | 0.243418 | 4.881 | –8.107 | –3.226 |
| B37-B | 0.591966 | 6.883 | –9.739 | –2.856 |
| B37-C | 0.528393 | 6.271 | –8.947 | –2.676 |
| B54-A | 0.214331 | 4.710 | –7.962 | –3.252 |
| B54-B | 0.612923 | 7.043 | –9.917 | –2.874 |
| B54-C | 0.51732 | 6.266 | –9.013 | –2.747 |
| B58-A | 0.207357 | 4.674 | –7.938 | –3.264 |
| B58-B | 0.582856 | 6.880 | –9.795 | –2.915 |
| B58-C | 0.511881 | 6.242 | –9.001 | –2.759 |
The results of natural electron configuration and AIM analysis were confirmed by spin density distribution analysis. Spin density is one of the diagnostic tools to determine the nature of the interaction between NO2 and C30B15N15. Spin density distributions of nine studied NO2-C30B15N15 complexes are visualized using GaussView software [29]. The visualized spin density distributions presented in Fig. 5. For all 9 studied NO2-C30B15N15 complexes, the spin densities are located somewhere other than the B-NO2 units. In the NO2-C30B15N15 complexes, where the gas molecule interacted with B37 and B54 of C30B15N15 heterofullerene, spin densities on the B-NO2 units not observed. For adsorption of NO2 through B58 of C30B15N15 heterofullerene, spin density distributions on the B-NO2 units are very small. Spin density distributions of 9 studied NO2-C30B15N15 complexes show that the interactions between NO2 and C30B15N15 are covalent in nature.

Visualized spin density distribution of NO2-C30B15N15 complexes.
4 Conclusions
The interaction between NO2 and C30B15N15 with different orientation of adsorbed gas investigated on the basis of density functional theory calculations. The results imply that NO2 can be chemically adsorbed on the exterior surface of C30B15N15 heterofullerene with a charge transfer from adsorbent to adsorbed gas. The results of NBO (natural electron configuration of adsorbing B atom), AIM analysis and spin density distributions revealed that the interaction of NO2 and C30B15N15 in all studied complexes are covalent in nature. From a qualitative aspect, all consequences are not dependent on the orientation of adsorbed gas.
Acknowledgements
The financial support of this work by the Iranian Nanotechnology Initiative is gratefully acknowledged. E. Zahedi expresses his gratitude to the Islamic Azad University of Shahrood.