

1 Abbreviations
DFT
Density Functional Theory
DPPH1,1-diphenyl-2-picrylhydrazyl
TBATFBtetrabutylammonium tetrafluoroborate
OTf = OSO2CF3trifluoromethanesulfonate
dppe1,1-Bis(diphenylphosphino)ethane
MLCTMetal Ligand Charge Transfer
LMCTLigand Metal Charge Transfer
GIAOGauge Independent Atomic Orbital
TMSTetrametylsilane
SVPSplit-Valence plus Polarization
TZVPTriple-Zeta Valence plus Polarization
2 Introduction
The use of cyclopalladated complexes as potential chemotherapeutics is an active area of research. These types of compounds are noted for their pharmacological properties, particularly as antibacterial [1–7], antifungal [3,5], and antitumoral agents [1–5,7,8]. Free radicals and chemical species with one or more unpaired electrons are produced in normal or pathological cell metabolism in different circumstances. Environmental stresses cause the faster generation of free radicals than their degradation in the cell [9]. Free radicals play a crucial role in the development of tissue damage in various human diseases such as cancer, aging, neurodegenerative disease, malaria and arteriosclerosis, and pathological events in living organisms [10]. Antioxidants may have an important role in the prevention of these diseases. There is an increasing interest in the antioxidant effects of compounds, especially natural moieties [11–13]. Inorganic antibacterial materials have several advantages over traditionally used organic agents; for example, chemical stability, thermal resistance, safety to the use and long lasting action period [14]. Bacterial resistance is a major drawback in chemotherapy of infectious disease [15]. The emergence of bacterial resistance to antibiotics and its dissemination, however, are major health problems, leading to treatment drawbacks for a large number of drugs. Currently there has been increasing interest in the use of inhibitors of antibiotic resistance for combination therapy [15–17]. Antibacterial agents are co-administered with an inhibitor that deactivates the bacteria's resistance mechanism and increases the antibacterial agent's effectiveness.
The coordination and organometallic chemistry of stabilized phosphorus ylides have been investigated extensively and their ambidenticity explained in terms of a delicate balance between electronic and steric factors [18–23]. The non-symmetrical stabilized ylides derived from bisphosphines, viz., Ph2P(CH2)nPPh2 = C(H)C(O)R (n = 1, 2) (R = Me, Ph or OMe) [24] form an important class of such ligands which can exist in ylidic and enolate forms. These ligands can therefore engage in different types of bonding [25]. The P, C bonding mode (Scheme 1) had been previously observed for Rh(I), Pd(II), Pt(II), Hg(II) [24–35] and also, the dppe chelating mode with Pd(II) has been found before [36]. The remarkable change in reactivity arises from a subtle variation in the molecular electronic structure of the ylide due to the presence of additional keto stabilization.
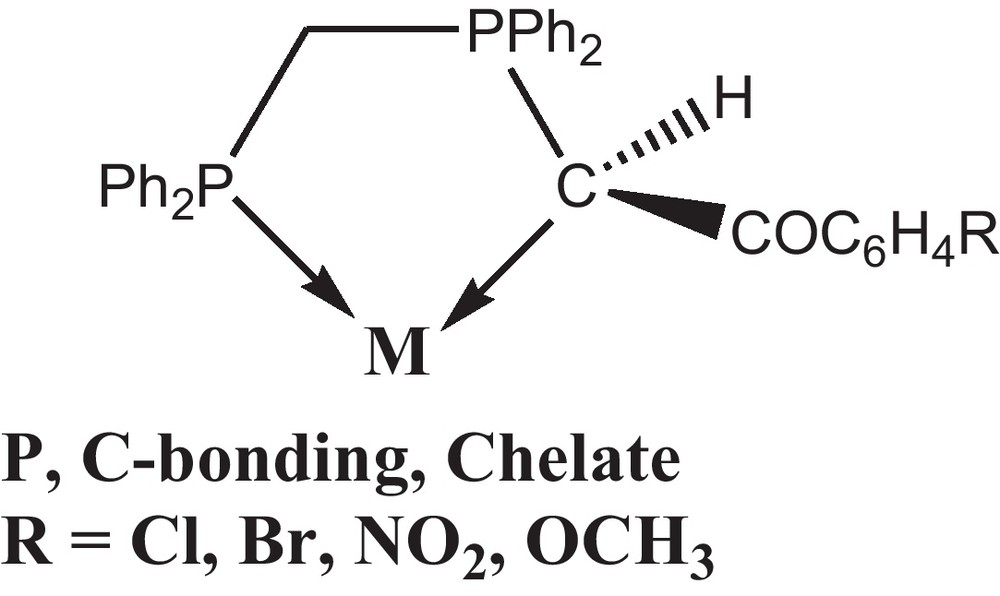
As part of this ongoing study we have chosen to investigate the bonding modes adopted by bifunctionalized ylide when coordinate to Pd(II). We have recently investigated the reaction chemistry of palladium(II) chloride complexes containing chelating non-symmetrical phosphorous ligands [25]. In this work, the dppe palladium halide complexes were usually reacted with OTf to extract the AgCl first. Then, the product was used as starting materials to react with non-symmetric phosphorus ylides to form chelated complexes. The results of these studies including the full characterization of the obtained complexes are presented. In addition, antioxidant and antibacterial activities of the synthesized complexes were assessed in vitro.
3 Experimental
3.1 Chemistry
In this study, four Pd(II) complexes with the dppe and the phosphorus ylides derived from the reaction between dppm and α-bromoketo derivatives, [(dppe)Pd(Ph2PCH2PPh2C(H)C(O)PhX)](OTf)2 (X = Br (C1), Cl (C2), NO2 (C3), OCH3 (C4)), were synthesized and investigated by physicochemical techniques. The reaction between [(dppe)Pd(OTf)2] and ylide (Y) in the 1:1 molar ratio readily occurred, leading to the new complexes [(dppe)Pd(Y)](OTf)2 (Scheme 2).
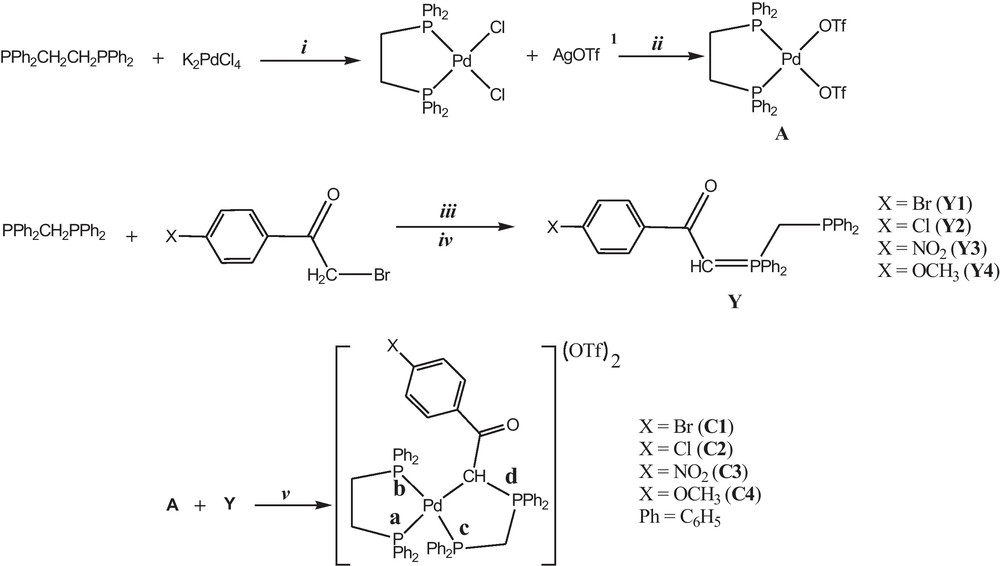
Synthetic route for preparation of Pd (II) complexes. In order to ease the visualization of the reactions, letter X refer to the substituents on para positions of the phenyl {X = Br (1), Cl (2), NO2 (3) and OCH3 (4)}. Reagents and conditions: (i) in ethanol, in the presence of hydrochloric acid and using Celite and reflux 8 h; (ii) in dichloromethane, using aluminum foil and Celite, 2 h at 25 °C; (iii) in chloroform, using dry nitrogen atmosphere, 2 h at 25 °C; (iv) in triethyl amine (0.5 ml) in toluene, 15 min in 25 °C; (v) in methanol, using dry nitrogen atmosphere, 12 h at 25 °C. 1 Silver trifluoromethanesulfonate.
3.2 Materials and physical measurements
The required chemicals were of analytical reagent grade and were purchased from Merck and Alderich. Melting points were measured on a SMPI apparatus and are reported without correction. Elemental analyses for C, H and N were performed using a Perkin–Elmer 2400 series analyzer. The analyses were repeated twice to check the accuracy of the analyzed data. Fourier transform IR spectra were recorded on a Shimadzu 435-U-04 spectrophotometer and samples were prepared as KBr pellets, in 200–4000 cm−1 region. UV-Vis spectra were recorded on a JASCO, V-670 Spectrophotometer, Japan, in the range 190–2700 nm. Mass spectra (electrospray) were recorded from a JEOL JMS 700 B/ES spectrometer. The thermo gravimetric analyses (TGA) were carried out in dynamic nitrogen atmosphere (20 ml min−1) with a heating rate of 10 °C min−1 in a platinum crucible using Mettler TA-4000/TG-50. The experiments were carried out on samples with an average mass of 10 mg. All electrochemical measurements were performed with a computer-controlled potentiostat, an Autolab electrochemical analyzer model PGSTAT30 (Eco Chemie, Utrecht, The Netherlands) and a standard three electrode cell consisting of an Au working electrode, a platinum wire auxiliary electrode and an Ag/AgCl (3 M KCl) reference electrode. The X-ray powder diffraction pattern was recorded over 2θ = 2–90° range using X-ray diffractometer model APD 2000. Radiation was provided by the copper anode (Kα, λ = 1.54056 Å) operated at 40 kV and 25 MA. NMR Spectra were recorded on a 90 MHz Jeol spectrometer (19F at 84.2 MHz and 31P at 36.26 MHz) or 400 MHz Bruker spectrometer (1H at 400.13 MHz) in CDCl3 as solvent at 25 °C. The splitting of proton resonances in the 1H and 31P NMR spectra are shown as: s = singlet, d = doublet, t = triplet and m = multiplet. Chemical shifts (ppm) are relative to internal TMS (δ = 0 (1H)), CFCl3 (δ = 0 (19F)) and external 85% phosphoric acid (δ = 0 (31P)).
3.3 Crystal structure determination and refinement
Suitable crystals for ligand Y2 was obtained from dichloromethane solution by slow evaporation of the solvent and mounted in random orientation on glass fibers. The X-ray intensity data were measured at 298 K on a Bruker SMART APEX CCD-based three-circle X-ray diffractometer system using graphite monochromated Mo Kα (λ = 0.71073 Å) radiation. The crystal structures were solved by direct methods and refined by using SHELXS-97 and SHELXL-97 crystallographic software packages [37–40]. All non-hydrogen atoms were refined anisotropically using reflections I > 2σ(I). Hydrogen atoms were located in ideal positions.
3.4 Computational details
The geometries of the complex C2, phosphorus ylide Y2 and dppe ligand in the gas phase were fully optimized using SVP and TZVP basis sets [41] at BP86 [42,43] level of theory. All calculations were performed using by Gaussian 03 [44] set of programs. Vibrational frequency analysis, calculated at the same level of theory, indicates that optimized structures are at the stationary points corresponding to local minima without any imaginary frequency. The 1H and 31P NMR shielding results were obtained at BP86 level of theory and using both the SVP and TZVP basis sets. Both the 1H and 31P isotropic shielding constants of the optimized mononuclear Pd (II) complex were calculated by using the gauge independent atomic orbital (GIAO) method [45,46].
3.5 Synthesis of phosphorus ylides Y1-4
General procedure [25]: Bis(diphenylphosphino)methane (dppm) (0.2 g, 0.52 mmol) was dissolved in 5 ml of chloroform and then a solution (5 ml, CHCl3) of 4-bromophenacyl bromide (0.144 g, 0.52 mmol) was added dropwise. The resulting reaction mixture was stirred for 2 h at room temperature. Addition of 25 ml of diethyl ether, after the prescribed reaction time caused the precipitation of the phosphonium salts. The resulting phosphonium salts (0.3 g, 0.45 mmol) were treated with triethyl amine (0.5 ml) in toluene (15 ml). The triethyl amine hydrobromide produced was filtered off and the toluene layer concentrated to 5 ml which upon further addition of petroleum ether (25 ml) resulted in the precipitation of the ligands as free-flowing solids. The product was collected and dried under vacuum (Scheme 2).
3.6 Synthesis of [Pd(dppe)Cl2] (M1)
General procedure: to a solution of the Bis(diphenylphosphino)ethane (dppe) (1 g, 2.5 mmol) in 20 ml ethanol was added to a stirred suspension of finely divided K2PdCl4 (0.81 g, 2.5 mmol) in 10 ml concentrated hydrochloric acid. The mixture were refluxed with stirring for eight hours and cooled to give precipitates of the product (1.6 g, 90%) (Scheme 2). 1H NMR (CDCl3): δ (ppm) 7.3–7.8 (m, 20H, C6H5), 1.82 (m, 4H, CH2). 31P NMR (CDCl3): δ (ppm) 66.7 (s, PPh2). Anal. Calc. for C26H24Cl2P2Pd: C, 54.24; H, 4.20, Found: C, 54.12; H, 4.11%.
3.7 Synthesis of [Pd(dppe)(OTf)2] (M2)
To a solution of complex M1 (0.57 g, 1 mmol) in CH2Cl2 (20 ml) was added AgOTf (0.51 g, 2 mmol). The reaction mixture was protected from room light with an aluminum foil and stirred for 2 h at room temperature. The solution was then filtered through dry Celite and the solvent was evaporated under reduced pressure. The residue was washed with diethylether (2 × 10 ml) and dried under vacuum (Scheme 2). Product [Pd(dppe)(OTf)2] was obtained as a white powder (0.67 g, 82%). 1H NMR (CDCl3): δ (ppm) 7.3–7.8 (m, 20H, C6H5), 1.94 (m, 4H, CH2). 31P NMR (CDCl3): δ (ppm) 60.1 (s, PPh2). 19F NMR (CDCl3): δ (ppm) 67.5 (s, CF3). UV–vis (λmax, nm): 262 (intra-ligand transition, phenyl group), 324 (MLCT). Anal. Calc. for C28H24F6O6P2PdS2: C, 41.88; H, 3.01. Found: C, 41.70; H, 3.11%.
3.8 Synthesis of Pd complexes
3.8.1 Synthesis of [(dppe)Pd(Y1)](OTf)2 (C1)
General procedure: to a [Pd(dppe)(OTf)2] (0.4 g, 0.5 mmol) methanol solution (10 ml), a solution of non-symmetric phosphorus ylides (Y1) (0.29 g, 0.5 mmol) (10 ml, CH3OH) was added dropwise. The resulting solution was stirred for 12 h at room temperature and then concentrated to a 2 ml in volume and treated with diethylether (2 × 10 ml) to give a final product. The product was collected and dried under vacuum (Scheme 2). White solid (0.58 g, 84%). M.p. 225–227 °C. 1H NMR (CDCl3): δ (ppm) 6.91–7.77 (m, 44H, C6H5), 2.66 (m, 2H, CH2(dppe)), 2.76 (m, 2H, CH2(dppe)), 4.82 (br, 1H, CH(methine)), 4.63 (m, 2H, CH2(ylide)). 31P NMR (CDCl3): δ (ppm) 55 (m, Pa), 55.1 (ddd, Pb, 2J(PbPc) = 355.1 Hz, 3J(PbPa) = 19.73 Hz, 3J(PbPd) = 11.96 Hz), 19.15 (ddd, Pc, 2J(PcPb) = 354.9 Hz, 2J(PcPa) = 58.1 Hz, 2J(PcPd) = 27 Hz), 37.65 (ddd, Pd, 2J(PdPb) = 58 Hz, 2J(PdPc) = 20.14 Hz, 3J(PdPa) = 14 Hz). 19F NMR (CDCl3): δ (ppm) -78 (s, CF3). Selected IR data (KBr, cm−1, w = weak, m = medium, s = strong): ν = 997 (m, P-CH2), 820 (m, P-CH), 1616 (s, C = O). UV–vis (λmax, nm): 241 (intra-ligand transition, phenyl group), 257 (intra-ligand transition, C = O), 300 (LMCT, s→d), 325 (MLCT). ESI-MS: m/z = 1235.06 ((Calc. 1235), [(dppe)Pd-Y1]+), 1085.1 ((Calc. 1086), [(dppe)Pd-Y1]2+). Anal. Calc. for C61H51BrF6O7P4PdS2: C, 52.92; H, 3.71. Found: C, 52.81; H, 3.65%.
3.8.2 Data for [(dppe)Pd(Y2)](OTf)2 (C2)
White solid (0.53 g, 80%). M.p.: 230–232 °C. 1H NMR (CDCl3): δ (ppm) 6.92–7.78 (m, 44H, C6H5), 2.67 (m, 2H, CH2(dppe)), 2.81(m, 2H, CH2(dppe)), 4.93 (m, 1H, CH(methine)), 4.52 (m, 2H, CH2(ylide)). 31P NMR (CDCl3): δ (ppm) 55.2 (m, Pa), 55.0 (ddd, Pb, 2J(PbPc) = 357.2 Hz, 3J(PbPa) = 20.81 Hz, 3J(PbPd) = 13.0 Hz), 19.53 (ddd, Pc, 2J(PcPb) = 355.7 Hz, 2J(PcPa) = 60.2 Hz, 2J(PcPd) = 26.8 Hz), 37.71 [ddd, Pd, 2J(PdPb) = 60.5 Hz, 2J(PdPc) = 20.5 Hz, 3J(PdPa) = 13.9 Hz). 19F NMR (CDCl3): δ (ppm) -77.9 (s, CF3). Selected IR data (KBr, cm−1): ν = 998 (m, P-CH2), 821 (m, P-CH), 1616 (s, C = O). UV–vis (λmax, nm): 245 (intra-ligand transition, phenyl group), 260 (intra-ligand transition, C = O), 310 (LMCT, s→d), 330 (MLCT). ESI-MS: m/z = 1191.2 ((Calc. 1191), [(dppe)Pd-Y1]+), 1041.1 ((Calc. 1040), [(dppe)Pd-Y1]2+). Anal. Calc. for C61H51ClF6O7P4PdS2: C, 54.68; H, 3.84. Found: C, 54.52; H, 3.66%.
3.8.3 Data for [(dppe)Pd(Y3)](OTf)2 (C3)
White solid (0.5 g, 75%). M.p.: 240–242 °C. 1H NMR (CDCl3): δ (ppm) 6.95–7.80 (m, 44H, C6H5), 2.6 (m, 2H, CH2(dppe)), 2.8 (m, 2H, CH2(dppe)), 5.22 (m, 1H, CH(methine)), 4.53 (m, 2H, CH2(ylide)). 31P NMR (CDCl3): δ (ppm) 60.9 (m, Pa), 59.02 (ddd, Pb, 2J(PbPc) = 356.2 Hz, 3J(PbPa) = 18.03 Hz, 3J(PbPd) = 13.52 Hz), 23.34 (ddd, Pc, 2J(PcPb) = 355.3 Hz, 2J(PcPa) = 60.4 Hz, 2J(PcPd) = 24.76 Hz), 40.94 (ddd, Pd, 2J(PdPb) = 60.1 Hz, 2J(PdPc) = 18.81 Hz, 3J(PdPa) = 13.8 Hz). 19F NMR (CDCl3): δF (ppm) -78 (s, CF3). Selected IR data (KBr, cm−1): ν = 998 (s, P-CH2), 827 (m, P-CH)], 1629 (s, C = O)]. UV–vis (λmax, nm): 245 (intra-ligand transition, phenyl group), 255 (intra-ligand transition, C = O), 300 (LMCT, s→d), 340 (MLCT). ESI-MS: m/z = 1202.1 ((Calc. 1200), [(dppe)Pd-Y1]+), 1050.06 ((Calc. 1051), [(dppe)Pd-Y1]2+). Anal. Calc. for C61H51F6NO9P4PdS2: C, 54.25; H, 3.81. Found: C, 54.69; H, 3.72%.
3.8.4 Data for [(dppe)Pd(Y4)](OTf)2 (C4)
White solid (0.57 g, 86%). M.p.: 212–214 °C. 1H NMR (CDCl3): δ (ppm) 6.5–7.82 (m, 44H, C6H5), 2.72 (m, 2H, CH2(dppe)), 2.96 (m, 2H, CH2(dppe)), 4.71 (m, 1H, CH(methine)), 4.51 (m, 2H, CH2(ylide)), 3.67 (s, 3H, CH3). 31P NMR (CDCl3): δ (ppm) 54.6 (m, Pa), 55.16 (ddd, Pb, 2J(PbPc) = 359.18 Hz, 3J(PbPa) = 22.0 Hz, 3J(PbPd) = 11.82 Hz), 19.31 (ddd, Pc, 2J(PcPb) = 359.15 Hz, 2J(PcPa) = 66.0 Hz, 2J(PcPd) = 21.28 Hz), 37.67 (ddd, Pd, 2J(PdPb) = 65.3 Hz, 2J(PdPc) = 20.2 Hz, 3J(PdPa) = 12.9 Hz). 19F NMR (CDCl3): δF (ppm) -78.3 (s, CF3). Selected IR data (KBr, cm−1): ν = 997 (m, P-CH2), 825 (s, P-CH), 1594 (s, C = O). UV–vis (λmax, nm): 240 (intra-ligand transition, phenyl group), 265 (intra-ligand transition, C = O), 310 (LMCT, s→d), 350 (MLCT). ESI-MS: m/z = 1035.2 ((Calc. 1036), [(dppe)Pd-Y1]+), 1185.2 ((Calc. 1185), [(dppe)Pd-Y1]2+). Anal. Calc. for: C, 55.76; H, 4.08. Found: C, 55.62; H, 4.06%.
3.9 Biological studies
3.9.1 DPPH radical scavenging assay
In order to determine the radical scavenging ability, the method reported by Mensor et al. [47] was used. Briefly, 0.3 mM alcohol solution of DPPH (1 ml) was added to samples (2.5 ml) containing different synthesized compounds. The samples were first kept in a dark place at room temperature and their absorbance was recorded at 517 nm after 30 min. The antiradical activity (AA) was determined using the following formula:
Blank samples contained 1 ml methanol + 2.5 ml from various concentrations of synthesized compounds; control sample containing 1 ml of 0.3 mM DPPH + 2.5 ml methanol. The optic density of the samples, the control and the empty samples were measured in comparison with methanol. A synthetic antioxidant, Ascorbic acid was used as positive controls. Experiments were carried out triplicate.
3.9.2 Antibacterial activity
The potential antibacterial effects of the complexes were investigated by disc diffusion method against two strains of Gram-positive bacteria, namely Bacillus cereus (PTCC 1247), Staphylococcus aureus (Wild) and two strains of Gram negative bacteria, namely Proteus vulgaris (PTCC 1079), and Serratia marcescens (PTCC 1111) [48]. The complexes were dissolved in hexane to a final concentration of 1 mg/ml and then sterilized by filtration using 0.45 μm Millipore. All tests were carried using 10 ml of suspension containing 1.5 × 108 bacteria per millilitre and spread on nutrient agar medium (NA). Negative controls were prepared by using hexane. Gentamycin and Penicillin were used as positive reference standards.
3.9.3 Statistical analysis
All data, for both antibacterial and antioxidant activity tests, are the average of triplicate analyses. Analysis of variance was performed by Excel and SPSS procedures. The statistical analyses were performed using Student's t-test, and P value < 0.05 was regarded as significant.
4 Results and discussion
4.1 FT-IR and 19F NMR studies
The proclivity of the OTf ligand to be replaced by nucleophiles is further shown by some observations indicating that in M2 the poorly coordinated OTf is continuously being changed with the phosphorus ylides. Fluorine NMR (measured in CDCl3) shows a weak peak for the trifluoromethanesulfonate ion in near 67.5 ppm in case of the complex M2 (Supplementary material, Fig. S1). 19F NMR spectroscopy in room temperature reveals a single resonance due to OTf at −77.9 ppm in complex C2 (Fig. S2), which indicates that the trifluoromethanesulfonate is a counter ion [49–51].
The IR spectrum of the ligands (Y1-Y4) and their complexes showed a band at ∼1500 cm−1 assigned to the carbonyl group. The characteristic band at 850 − 900 cm−1 of the P–CH group of the phosphorus ylide and its complexes are assigned to P–C vibration (Table 1). The ν (CO), which is sensitive to complexation, occurs around 1600 cm−1 in the parent ylides [52,53]. Coordination of the ylide through carbon causes an increase in the ν (CO), while, when O-coordination occurs, a lowering in the value of the ν (CO) would be expected [54]. Thus the IR absorption bands for the complexes at higher frequencies indicate that C-coordination has occurred [33].
Comparison 31P{1H} NMR and IR bands of the phosphorus ylides and chelated complexes.
| Complexes & Ligands | 31P{1H} NMR, ppm | J (PP), Hz | FT-IR (cm−1) | |||||||||
| δPa | δPb | δPc | δPd | J ab | J ac | J ad | J cd | J bd | J cb | ν (CO) | ν (PCH) | |
| Y1 | −30.0 | 11.4 | 63.7 | 1502 | 881 | |||||||
| C1 | 55.0 | 55.1 | 19.1 | 37.6 | 19.7 | 58.1 | 14.0 | 27.0 | 11.9 | 355.1 | 1616 | 820 |
| Y2 | −29.9 | 11.3 | 63.1 | 1503 | 846 | |||||||
| C2 | 55.2 | 55.0 | 19.5 | 37.7 | 20.8 | 60.2 | 13.9 | 26.8 | 13.0 | 355.7 | 1616 | 821 |
| Y3 | −30.4 | 11.5 | 62.5 | 1524 | 862 | |||||||
| C3 | 60.9 | 58.8 | 23.8 | 41.0 | 18.0 | 60.4 | 13.8 | 24.7 | 13.5 | 355.3 | 1629 | 827 |
| Y4 | −29.6 | 11.1 | 62.4 | 1507 | 876 | |||||||
| C4 | 55.0 | 55.4 | 19.3 | 37.8 | 22.0 | 66.0 | 12.9 | 21.3 | 11.8 | 359.2 | 1594 | 825 |
4.2 31P NMR studies
In the 31P{1H} NMR spectra of the Ph2PCH2PPh2C(H)C(O)PhX (Y1–Y4) the signal due to PCH (Pd) and PCH2 (Pc) appears as a two doublet in ∼ 11 and -30 ppm, respectively (Fig. S3). Also, the 31P chemical shift of M1 due to Pa and Pb appears as singlet in 66 ppm (Fig. S4). The 31P NMR spectrum shows an interesting pattern of signals which allows assigning definitely the structure of these complexes (C1-C4). The four P atoms in these complexes are chemically and magnetically non-equivalent (Scheme 2); therefore, the 31P {1H} NMR spectrum exhibits four batch-peak (Fig. 1). The upfield peak (δP ≈ 55–60, m), (Δδ ≈ −10 ppm) is assigned to the P atom (Pa) of the bidentate ligand (dppe), which is directly attached to the Pd centre in the front of CH group and thus shielded. The 3JPbPa coupling constant of ∼20 Hz lies within the typical values of similar systems [55–57]. Following, the 2JPcPa and 3JPdPa coupling constant are ∼60 and ∼13 Hz, respectively [34]. The upfield peak (δP ≈ 55–59, ddd), (Δδ ≈ −8 ppm) is assigned to the P atom (Pb) of the dppe, which is directly attached to the Pd centre in the front of PCH2 group (Pc) and thus shielded (Table 1). The extremely strong coupling with the P atom (Pb) of the dppe ligand (2JPbPc ≈ 355 Hz) can only be rationalized in the context of a trans arrangement between Pb and Pc [56–64]. The 3JPbPd coupling constant is ∼12 Hz. Two down-field peaks are assigned to two P atoms of the chelate phosphorus ylide. The ylidic P atom (Pd) of the phosphorus ylide is not directly coordinated to the metal centre, thus coupling with Pc is weak (2JPdPc ≈ 24 Hz) as expected [34]. Coupling with Pb (3JPdPb ≈ 12) and Pa (3JPdPa ≈ 13 Hz) gives rise to a downfield ddd-peak (δP ≈ 37–41, ddd), (Δδ ≈ +30 ppm). The chemical shifts for the two non-equivalent P-atoms of the ambidentate P,CH-ligand are assigned on the basis of published NMR data for these complexes [25]. Finally, the downfield peak (δP ≈ 19–24, ddd), (Δδ ≈ +50 ppm) is assigned to PCH (Pc).
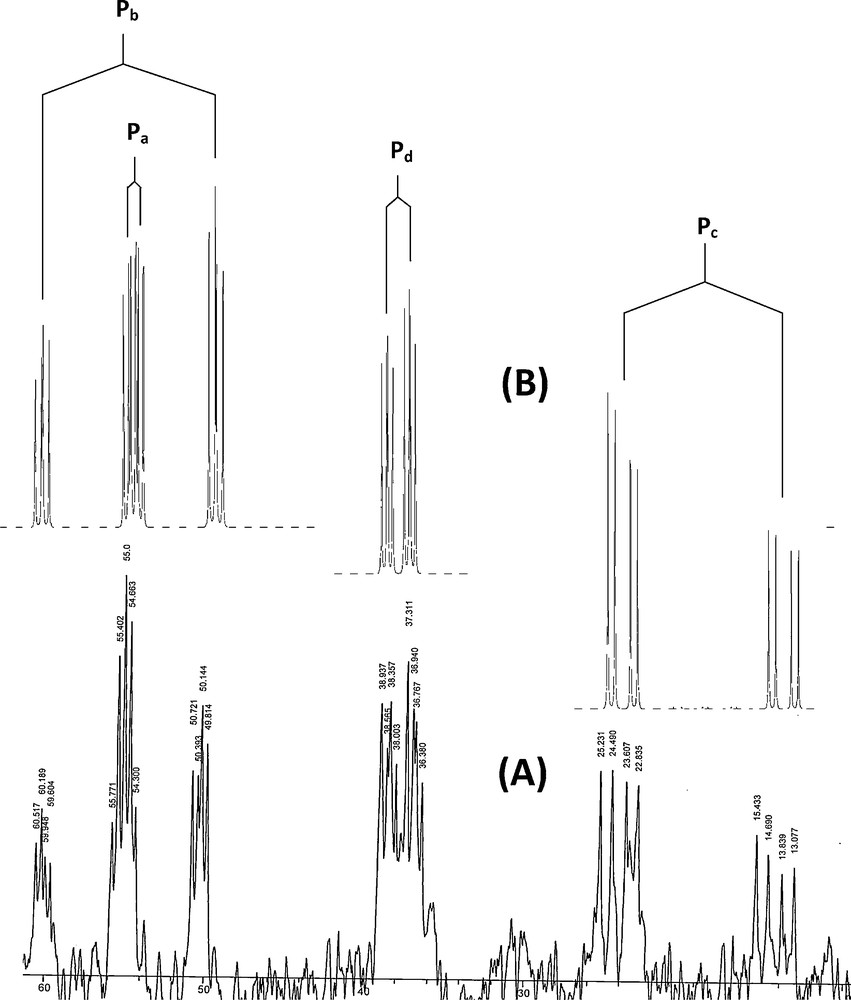
The 31P{1H} NMR spectrum of complex C1. (A): real spectrum; (B): simulated spectrum.
The significant downfield shift of the signal from that of the free ylide is in agreement with the C-bonding of the ylides. The coordination via the phosphine moiety is also clearly evidenced from the strong downfield shifts of the signal due to PPh2 group when compared to that of the same signal in the free ylide [33,58].
4.3 1H NMR studies
The d8 distorted square planar palladium(II) complexes are all diamagnetic and give resolved NMR spectra which confirm the nature of the binding [65]. Regarding the 1H NMR spectrum of these complexes, the phenyl protons of the dppe ligand and phosphorus ylide are observed in the aromatic region of the spectrum, between 6.5–8.0 ppm [25]. According to the published data, the 1HNMR spectrum of the palladium(II) complexes was consistent with coordination of the phosphorus ylide to the metal through the CH and Pc atoms. In the 1H NMR spectrum, the signal due to the methene proton for complex C1 is broad [25]. Similar behavior was observed earlier in the case of ylide complexes derived from PtCl2 [66], whereas the complexes C2-C4 showed a multiple peak in ∼4.9 ppm due to the methene proton. The expected lower shielding of the 1H nuclei for the PC(H) group upon complexation in the case of C-coordination was observed in their corresponding spectra [67]. All complexes showed a multiple peak in near 2–4 ppm due to CH2 groups of the dppe ligand (e.g. complex C4, Fig. S5). The CH2 protons in all complexes of the phosphorus ylide appear as a multiplet (m) δ ≈ 4.5 ppm (Fig. S5). A singlet appeared at 3.67 ppm corresponding to methyl group of protons in complex C4.
4.4 ESI mass spectrometry
The positive ion electrospray-mass spectra (ESI+-MS) of the complexes C1–C4 were recorded as very dilute THF solution. The molecular ion was observed in the ESI mass spectra of all complexes. The phosphorus ylide Y1 has a molecular ion peak at m/z 583. The mass spectrum of complex C1 was characterized by the appearance of the fragment at m/z 1085.1 owing to the removal of two OTf group and [(dppe)Pd-Y1]2+ was formed. A peak at m/z 1235.06 related to the molecular ion corresponds to elimination of one OTf ions to give [(dppe)Pd-Y1]+OTf. Similar fragmentation patterns are observed for complexes C1–C4 and closely matched to what is expected and clearly confirmed the formation of these compounds. Modeling pattern of the complex C1 and C4 are presented in Fig. 2 and Fig. S6, respectively.
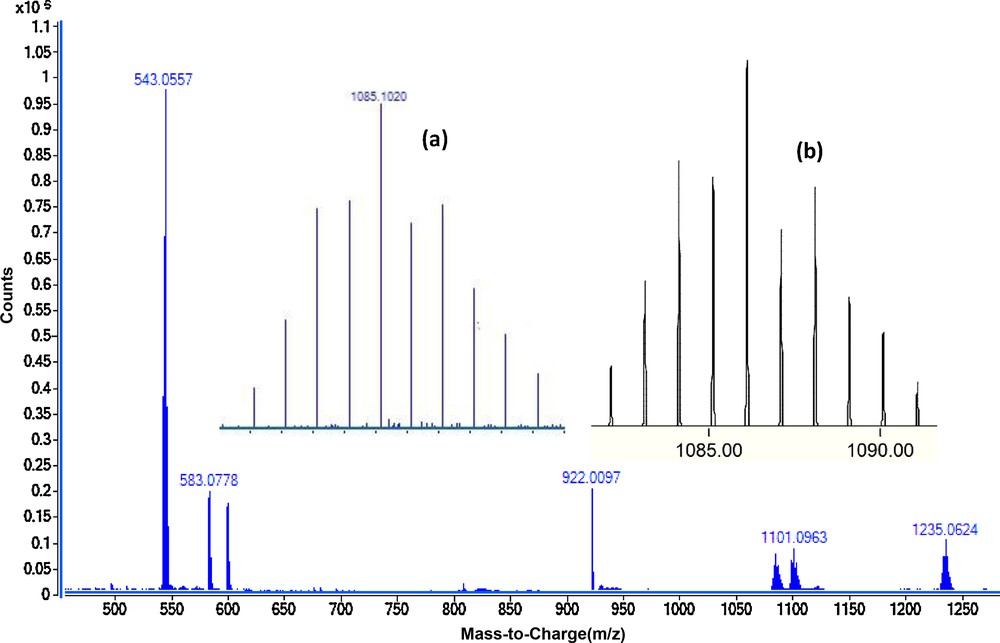
ESI-MS (dilute THF) of complex C1. The inset spectrum shows the overlaid experimental (a) and calculated (b) isotope patterns for [Pd(dppe)(Y1)]2+.
4.5 Electronic absorption
All palladium complexes have been found to be diamagnetic indicating a +2 oxidation state for palladium. The electronic spectra of the complexes have been recorded in solid state and displayed three bands in the region around 200–500 nm. The electronic spectra of the phosphorus ylides as a ligand showed spectral bands due to π→π* and n→π* transitions (Fig. S7). On complexation these bands are shifted (Fig. S7). The strong charge transfer transitions may interfere and prevent the observation of all the expected bands [68–70]. The strong bands ∼400 nm is assignable to a combination of metal ligand charge transfer (MLCT) and d-d [68,71]. The broad band at 330–370 nm (28,700 cm−1) is assignable to combination of phosphor/metal charge transfer (LπMCT) and d-d bands. The broad band that appeared around 470 nm (21,250 cm−1) has been reported as due to both distorted square planar and undistorted square planar Pd(II) complexes [72,73]. The bands that appeared around 250, 350 and 450 nm have been assigned to intra-ligand transition, [74,75] LMCT [74–76] and MLCT [77,78], respectively. The two bands between 230 and 260 nm (39,000–42,200 cm−1) in the Pd(II) complexes are assigned to ligand centered transitions (π-π* and n-π*).
The band near 31,740 cm−1 in complex C1 is spin-allowed transition; a1g(dx2−y2) − b1g(dx2−y2) i.e. 1A1g → 1B1g. The broad bands between 340 and 360 nm (27,700–29,400 cm−1) in the palladium complexes are assignable to a combination of MLCT and eg(dyz,dxz) − b1g(dx2−y2), i.e. 1A1g → 1Eg bands. Therefore, the electronic spectra of Pd (II) complexes are indicative of distorted square planar geometry [79–81].
4.6 Electrochemical studies
Electrochemical studies of M2, phosphorus ylides as ligand (Y1-Y4) and Pd (II) complexes (C1-C4) have been carried out by cyclic voltammetry (CV) in dichloromethane solution, containing 0.1 M tetrabutylammonium tetrafluoroborate (TBATFB) as supporting electrolyte using Au working electrode and Pt wire counter electrode. All the potentials were referenced to Ag/AgCl reference electrode.
Cyclic voltammograms of 1 mM of M2, Y3 and C3 in dichloromethane, containing 0.1 M TBATFB at a scan rate of 100 mV/s is shown in Fig. 3. For the complex M2 the cathodic potential peak corresponds to the redox pair of Pd(II)/Pd(I) appeared at −0.65 V vs. Ag/AgCl. That reduction was due to one electron transfer process. When the potential was scanned further in the cathodic direction, a second cathodic potential peak at −1.12 V was observed, which corresponds to the redox pair of Pd(I)/Pd(0). There were no anodic peaks observed on the reverse scan [82]. This behavior is similar to that reported previously in cyclic voltammetric study of various types of Pd(II) complexes in aprotic solvents [82–84]. This irreversibility may be due to the reaction of a Pd(0) complex with adventitious O2 or other components of the solution. Another cathodic peak observed at −0.45 V is corresponding to the reduction of dppe ligand.
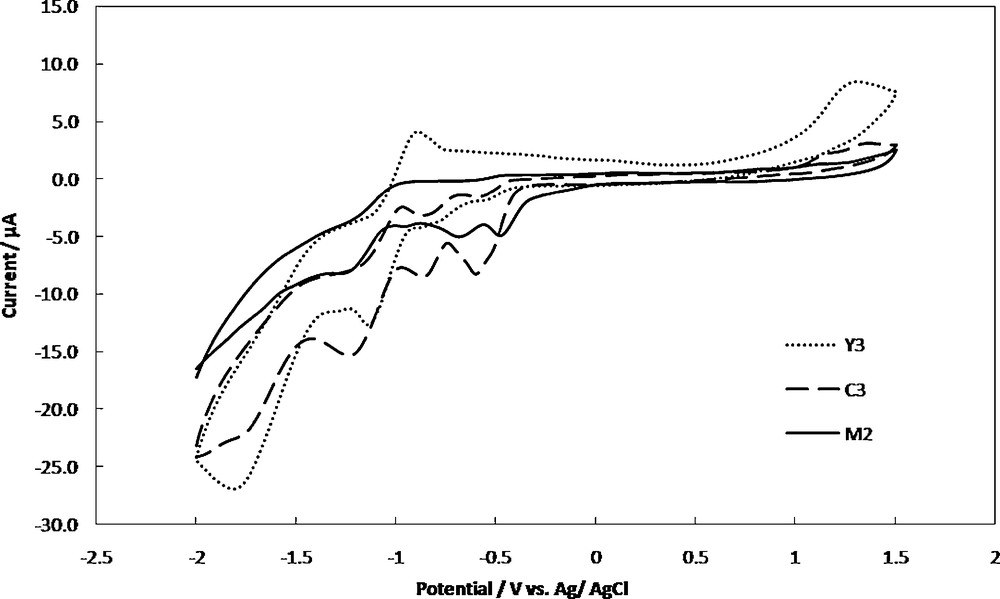
Cyclic voltammograms of 1 mM of M2, Y3 and C3 in dichloromethane containing 0.1 M TBATFB as supporting electrolyte, at the Au electrode and scan rate of 100 mV/s.
For Y3, the cyclic voltammogram displayed two irreversible and quasi reversible reduction peaks at −1.65 V and −1.10 V, respectively. The first peak could be assigned to a four-electron reduction of nitro group generating the hydroxylamine derivative according to:
The second cathodic peak assigned to the further reduction of the hydroxylamine to p-amino derivative [7,85].
In addition, the CV of the C3 exhibited an irreversible reduction peak at −1.60 V corresponding to the reduction of nitro group. The reduction process is shifted towards lower potential revealed an enhancement of the reduction process in the presence of palladium [7]. The disappearance of the second reduction peak of nitro group indicated the participation of the hydroxylamine group in the complex formation.
The quasi reversible reduction peaks at −0.82 V and −1.18 V can be assigned to the formation of Pd(I) from Pd(II) and formation of Pd(0) from Pd(I), respectively. The irreversible cathodic peak at −0.55 V is corresponding to the reduction of dppe ligand. The reduction process is shifted toward more negative potential in the presence of nitro group. The existence of the cathodic peaks of Pd, NO2 and the dppe in the CV of the C3 complex, is an evidence for the synthesis of this complex. The cyclic voltammetric data of metal center for the other complexes are given in Table 2.
Electrochemical data of the palladium (II) complexes.
| Complex | Epc PdII-PdI (V) | Epc PdI-Pd0 (V) |
| C1 | −1.25 | −0.81 |
| C2 | −1.24 | −0.79 |
| C3 | −1.18 | −0.82 |
| C4 | −1.10 | −0.68 |
4.7 Thermal analyses
Thermal stability of the complexes was determined by TG/DTG studies from 25–1000 °C. The compounds were heated to 950–1000 °C, all relevant weight loss was completed by 800 °C. Discussing the simultaneous TG/DTG curves constructed for phosphorus ylide (Y1), one can observe two endothermic mass loss stages (supplementary material, Fig. S8). The 1st decomposition step at 80–710 °C is accompanied by a mass loss of 80.72% (calcd. 86.72%) may be attributed to the liberation of ylide without bromine atom. The decomposition ended at 797 °C with bromine as a final residue (obs. = 17.49%, calc. = 13.74%). The TG/DTG curves of C1 (Fig. S8) exhibit three decomposition steps maximized at 391, 615 and 797 °C. The first pronounced peak at 157–593 °C is accompanied by a mass loss amounting to 76.28% (calcd. 75.76%) may be attributed to the liberation of dppe + Y1 + CF3. The second decomposition stages at 593–707 °C involve the loss of O2. The decomposition ended at 797 °C with Pd + SO + SO3CF3 as a final residue (obs. = 18.40%, calc. = 21.92%).
For ylide (Y3), the degradation starts at 25 °C with four decomposition stages centered at 116, 285, 366 and 797 °C (Fig. 4a). The 1st and 2rd decomposition stages are attributed to the degradation of nitro group. The 3rd degradation step is due to the releasing of dppm (obs. = 68.50%, calc. = 70.20%). Decomposition ended at 797 °C with acetophenone as a final residue (obs. = 20.83%, calc. = 21.94%). The TG/DTG curves of complex C3 (Fig. 4b) exhibited three decomposition steps maximized at 369, 461 and 797 °C. The 1st pronounced peak at 190–439 °C is accompanied by a mass loss amounting to 67.23% (calcd. 66.78%) may be attributed to the liberation of dppe and one ylide (Y3) without NO2 group. The second thermal stage, centered at 439–652 °C, is due to the degradation of 3O2 with overall mass loss amounting to 21.93% (calcd. 26.40%) leaving two HSOCF3, one NO2 group and Pd as a final residue.
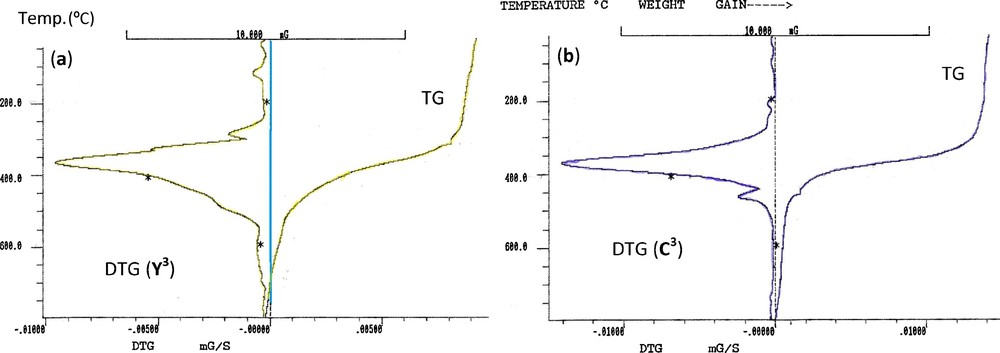
TG and DTG curves of (a) ylide Y3 (b) complex C3.
4.8 X-ray crystallography
Table 3 summarizes crystal and refinement data for Y2. The molecular structure is shown in Fig. 5. This compound crystallizes in a monoclinic space group P21/c. As shown, both the phosphine and ylidic components are devoid of steric restrictions and can interact with metal ions and reagents. In the molecule of the title ligand, Y2, the geometry around the P1 atom is nearly tetrahedral and the O atom is oriented cis to the P atom. The C(27)–C(26)–P(2) [115.3 (3)°] bond angle indicates a distorted trigonal arrangement about C(26). The P(1)-O [2.9104 (16) Å] distance is significantly shorter than the sum of the van der Waals radii of P and O (3.3 A), [86] indicating a strong intermolecular interaction between P+ and O−charge centers, which leads to the cis orientation. The P(2)–C(26) [1.711 (5) Å], C(27)–O(1) [1.267 (5) Å] and C(26)–C(27) [1.396 (7) Å] bond lengths within the ylidic fragment are comparable to those observed for the other monoketo ylides [87,88]. Thus, no steric or electronic effects due to the presence of –CH2PPh2 are anticipated. Also, in this molecule the bond lengths and angles are generally within normal ranges [33]. In ligand Y2 a combination of several weak and medium intermolecular interactions, determined the structural assembly in this compound (Fig. 6).
Crystal data and structure refinement details for Y1.
| Identification code | Y1 |
| Empirical formula | C33H28ClOP2 |
| Formula weight | 537.94 |
| Temperature (K) | 150 (2) |
| Wavelength (Å) | 0.71073 |
| Crystal system space group | Monoclinic P21/c |
| Unit cell dimensions (Å) | a = 13.4006 (6) |
| b = 18.6384 (9) | |
| c = 11.1601 (5) | |
| β = 100.973 (4)° | |
| Volume (Å3) | 2736.45 |
| Z, Calculated density (Mg/m3) | 4, 1.306 |
| Absorption coefficient (mm−1) | 0.282 |
| F (000) | 1124.0 |
| Crystal size (mm) | 0.65 × 0.30 × 0.13 mm |
| θ Range for data collection (°) | 3.28 to 25.00 |
| Limiting indices | −15 ≤ h ≤ 15 |
| −18 ≤ k ≤ 22 | |
| −13 ≤ l ≤ 11 | |
| Reflections collected/unique | 12500/4794 [R (int) = 0.0687] |
| Completeness | 99.6% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.964 and 0.838 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4794/252/395 |
| Goodness-of-fit on F2 | 1.119 |
| Final R indices [I > 2 σ (I)] | R1 = 0.0837, wR2 = 0.1683 |
| R indices (all data) | R1 = 0.1361, wR2 = 0.1871 |
| Largest diff. peak and hole (eÅ3) | 1.327 and −0.314 |
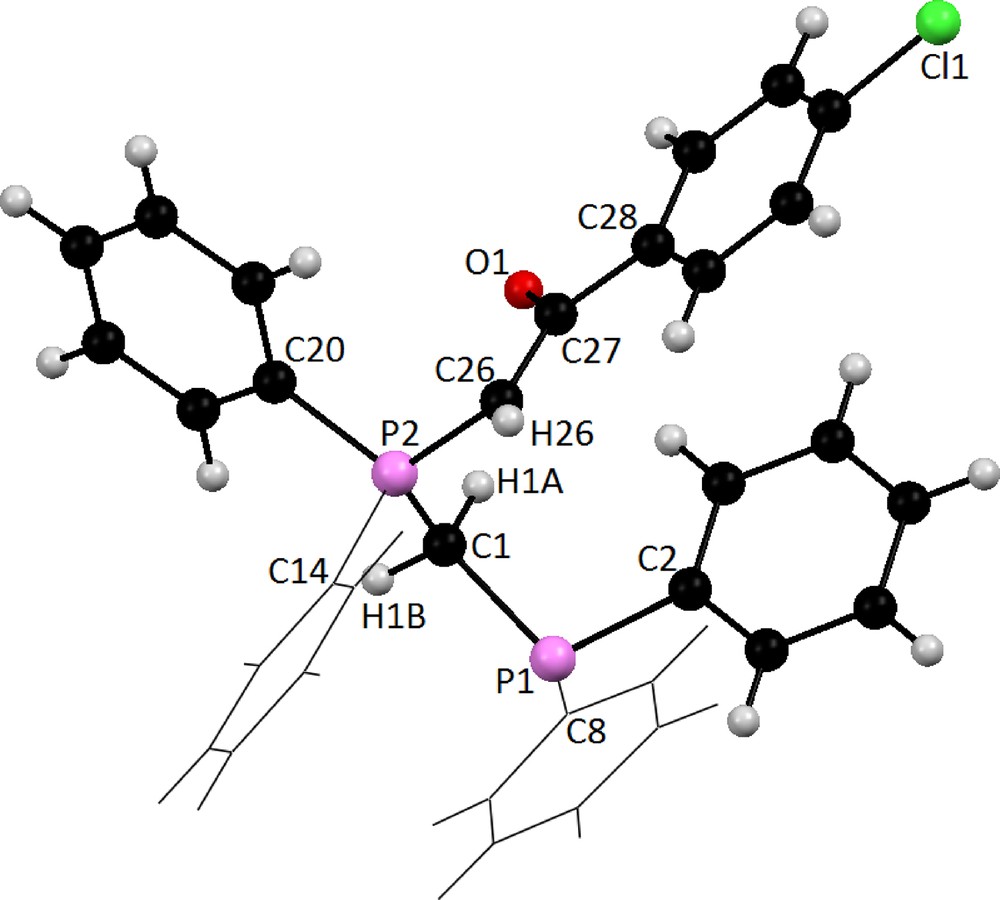
Molecular structure of Y2 showing the atom-labeling scheme.
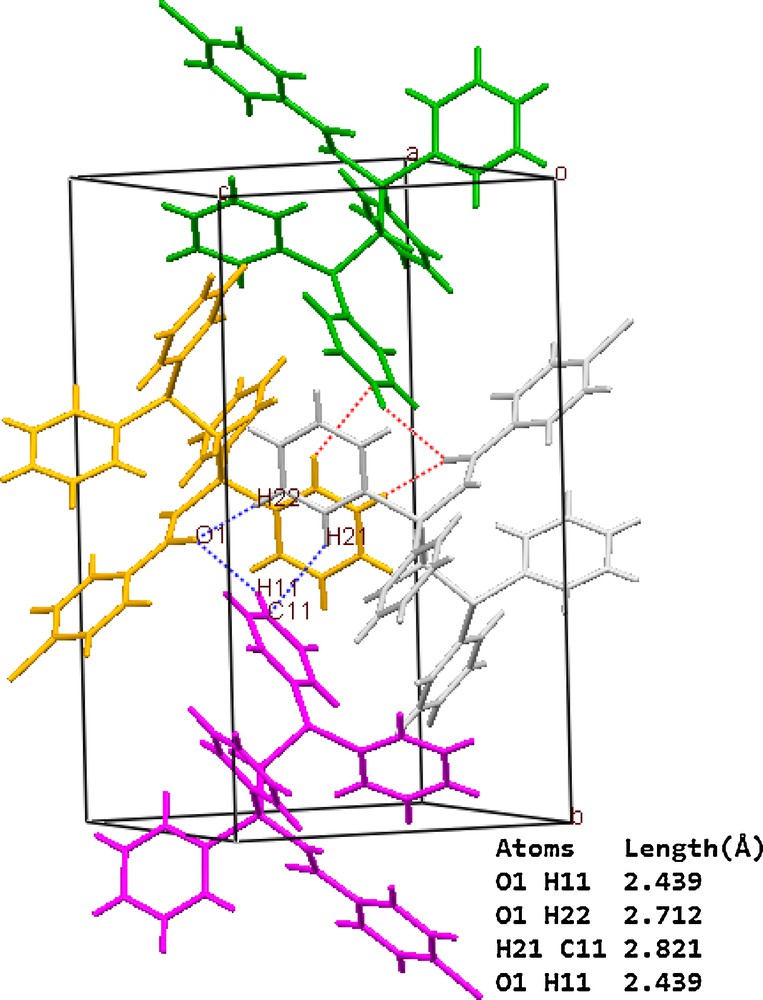
A representation of ligand Y2 packing showing the association of the adjacent molecules through C-Hphenyl…πphenyl, C-Hphenyl…Ocarbonyl interactions. Dotted lines represent the short contacts.
4.9 X-ray powder diffraction
Single crystals of the studied complexes could not be obtained, because the studied complexes are amorphous by nature. The X-ray powder diffraction patterns of the phosphorus ylide (Y3) and its complex as representative examples were recorded over 2θ = 5–90° in order to obtain an idea about the lattice dynamics of these compounds. A comparison of the obtained XRD patterns of Y3 and C3, throws light on the fact that each compound represents a definite compound with a distinct structure (Fig. S9). This identification of the compounds was done by the known method [89]. Such facts suggest that the prepared complexes are amorphous [90]. The X-ray powder diffraction pattern of Y3 showed a higher degree of crystallinity. The values of 2θ, interplanar spacing d (Å) and the intensities (%) of Y3 and its complex were tabulated in Table 4. Two peaks characterized to the Pd (II) moiety are also observed at 36.5° and 44.2° [91].
X-ray diffraction data of phosphorus ylide (Y3) and Pd complex (C3).
| Y3 | C3 | ||||
| Angle (2θ) | d-Value (Å) | Intensity (%) | Angle (2θ) | d-Value (Å) | Intensity (%) |
| 3.8 | 23.22 | 21.8 | 2.6 | 33.95 | 78.0 |
| 6.9 | 12.80 | 21.7 | 3.4 | 25.96 | 54.7 |
| 8.7 | 10.15 | 24.7 | 3.8 | 23.23 | 51.8 |
| 9.7 | 9.11 | 20.0 | 7.9 | 11.18 | 56.1 |
| 12.2 | 7.24 | 22.6 | 8.2 | 10.77 | 59.1 |
| 12.6 | 7.01 | 20.9 | 9.1 | 9.71 | 64.5 |
| 13.0 | 6.80 | 19.4 | 9.6 | 9.20 | 67.7 |
| 13.4 | 6.60 | 33.8 | 10.2 | 8.66 | 53.1 |
| 13.8 | 6.41 | 23.4 | 10.4 | 8.49 | 53.7 |
| 14.8 | 5.98 | 16.3 | 11.5 | 7.68 | 46.0 |
| 16.4 | 5.40 | 19.1 | 15.5 | 5.71 | 49.1 |
| 16.7 | 5.30 | 18.9 | 15.8 | 5.60 | 46.3 |
| 17.4 | 5.09 | 76.1 | 16.2 | 5.46 | 52.4 |
| 18.3 | 4.84 | 43.6 | 16.5 | 5.36 | 61.0 |
| 19.7 | 4.50 | 91.0 | 17.2 | 5.15 | 60.7 |
| 20.6 | 4.30 | 77.3 | 17.8 | 4.97 | 63.1 |
| 22.1 | 4.01 | 32.1 | 18.3 | 4.84 | 58.2 |
| 22.9 | 3.88 | 41.4 | 18.7 | 4.74 | 63.1 |
| 23.5 | 3.78 | 27.8 | 19.2 | 4.61 | 70.6 |
| 24.8 | 3.58 | 60.9 | 19.6 | 4.52 | 68.1 |
| 25.7 | 3.46 | 46.5 | 20.0 | 4.43 | 70.8 |
| 26.6 | 3.34 | 28.4 | 20.7 | 4.28 | 69.5 |
| 28.8 | 3.09 | 32.5 | 21.0 | 4.22 | 74.9 |
| 30.1 | 2.96 | 21.9 | 21.5 | 4.12 | 77.1 |
| 30.6 | 2.91 | 20.6 | 22.1 | 4.01 | 76.1 |
| 31.3 | 2.85 | 24.9 | 22.4 | 3.96 | 76.7 |
| 31.6 | 2.82 | 28.0 | 22.8 | 3.89 | 65.6 |
| 32.0 | 2.79 | 21.9 | 23.5 | 3.78 | 71.3 |
| 33.4 | 2.68 | 22.3 | 23.9 | 3.72 | 62.2 |
| 41.2 | 2.18 | 18.6 | 24.5 | 3.63 | 67.6 |
| 25.5 | 3.49 | 64.9 | |||
| 25.8 | 3.45 | 66.0 | |||
| 28.7 | 3.10 | 56.2 | |||
| 30.4 | 2.93 | 60.6 | |||
| 30.8 | 2.90 | 65.4 | |||
| 31.5 | 2.83 | 52.3 | |||
| 31.8 | 2.81 | 53.8 | |||
| 32.1 | 2.78 | 51.8 | |||
| 33.4 | 2.68 | 49.3 | |||
| 36.5 | 2.45 | 45.6 | |||
| 43.5 | 2.07 | 39.4 | |||
| 44.2 | 2.04 | 45.6 |
4.10 Theoretical studies
4.10.1 Geometry optimization
The optimized geometries of phosphorus ylide (Y2) and dppe ligand are depicted in Fig. S10 and the metal complex C2 is presented in Fig. 7. Representative selected calculated bond parameters for bidentate ligands Y2 and dppe along with the experimentally determined data are listed in Table 5. The calculated bond lengths and bond angles for both ligands are in good agreement with the corresponding experimental values. As can be seen in Fig. 7, the coordination sphere around the metallic center in complex C2 is made up of CHylide, Pylide and two Pdppe completing the distorted square planar geometry. The P(3)-Pd-P(4) and C(2)-Pd-P(5) bond angles are 168.3 and 174.6°, respectively, indicating that the four donor atoms are not coplanar (Table 6).
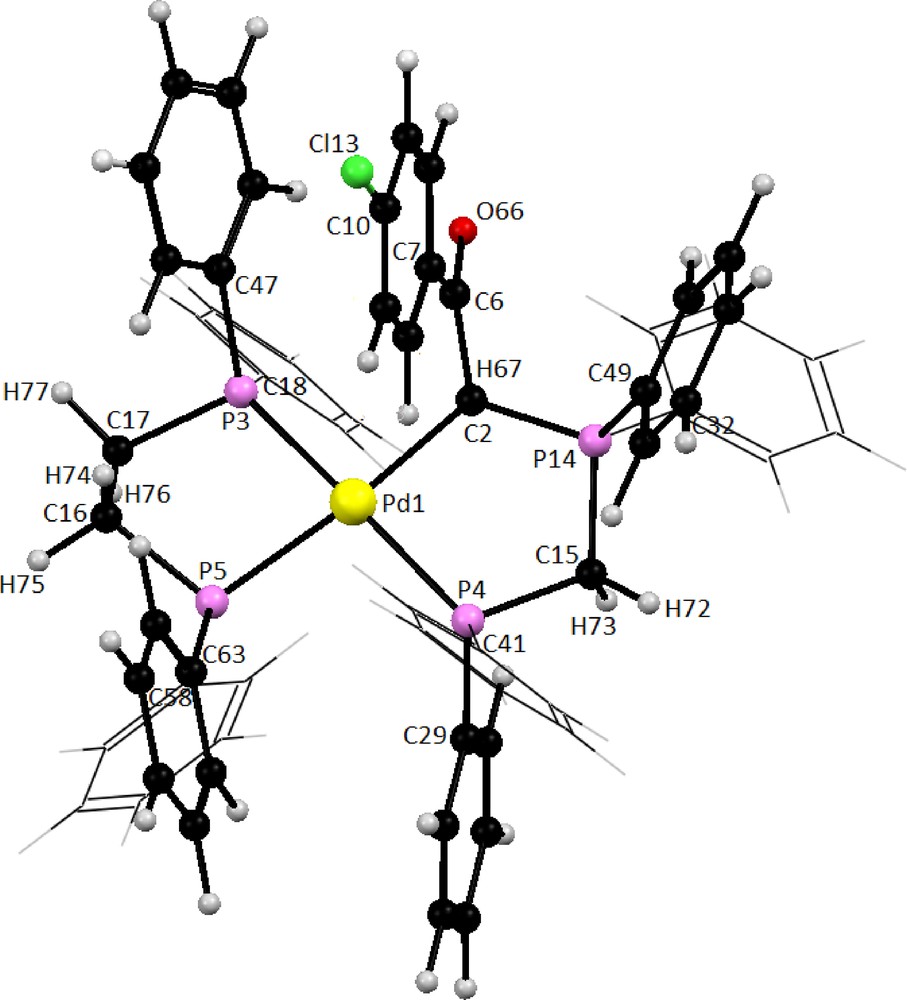
Optimized structure of complex cation in compound C2 at Bp86/TZVP level of theory.
Selected bond distances (Å) and angles (°) for phosphorus ylide Y2 and dppe.
| Y2 | dppe | ||||||
| Exp. | BP86/SVP | BP86/TZVP | Exp. [92] | BP86/SVP | BP86/TZVP | ||
| Bond distances | Bond distances | ||||||
| P2-C11 | 1.853 (4) | 1.907 | 1.896 | P1-C2 | 1.828 (4) | 1.893 | 1.881 |
| P2-C17 | 1.81 (2) | 1.860 | 1.847 | P1-C7 | 1.820 (3) | 1.862 | 1.850 |
| P2-C29 | 1.827 (4) | 1.874 | 1.860 | P1-C8 | 1.818 (4) | 1.854 | 1.842 |
| P10-C11 | 1.821 (4) | 1.852 | 1.842 | C2-C3 | 1.516 (4) | 1.530 | 1.528 |
| P10-C20 | 1.799 (5) | 1.847 | 1.832 | C2-H29 | 0.88 (3) | 1.112 | 1.102 |
| P10-C31 | 1.818 (5) | 1.840 | 1.829 | C2-H30 | 0.96 (3) | 1.112 | 1.102 |
| P10-C1 | 1.711 (5) | 1.742 | 1.728 | C8-C19 | 1.370 (7) | 1.417 | 1.409 |
| C11-H43 | 0.989 (4) | 1.110 | 1.101 | C8-C23 | 1.362 (6) | 1.415 | 1.407 |
| C11-H44 | 0.991 (4) | 1.110 | 1.100 | C19-C20 | 1.375 (10) | 1.404 | 1.396 |
| C1-C3 | 1.396 (7) | 1.418 | 1.413 | C20-C21 | 1.328 (11) | 1.407 | 1.399 |
| C1-H38 | 0.89 (4) | 1.098 | 1.088 | C21-C22 | 1.346 (10) | 1.405 | 1.397 |
| C3-O36 | 1.267 (5) | 1.270 | 1.266 | C22-C23 | 1.382 (8) | 1.406 | 1.399 |
| C3-C4 | 1.505 (6) | 1.513 | 1.509 | ||||
| C4-C9 | 1.385 (6) | 1.412 | 1.404 | Bond angles | |||
| C8-C9 | 1.385 (7) | 1.401 | 1.394 | C2-P1-C8 | 102.6 (3) | 103.479 | 103.691 |
| C7-C8 | 1.369 (8) | 1.407 | 1.397 | C2-P1-C7 | 100.1 (3) | 101.545 | 101.864 |
| C6-C7 | 1.378 (7) | 1.407 | 1.397 | C7-P1-C8 | 101.3 (3) | 103.283 | 103.518 |
| C5-C6 | 1.379 (8) | 1.403 | 1.396 | P1-C7-C28 | 119.0 (5) | 125.123 | 125.217 |
| C4-C5 | 1.394 (8) | 1.413 | 1.405 | P1-C7-C24 | 123.2 (5) | 116.351 | 116.358 |
| C7-Cl37 | 1.749 (5) | 1.755 | 1.751 | P1-C8-C23 | 116.7 (6) | 121.940 | 121.877 |
| P1-C8-C19 | 125.6 (6) | 119.423 | 119.522 | ||||
| Bond angles | P1-C2-C3 | 111.1 (4) | 118.898 | 118.829 | |||
| C17-P2-C29 | 102.6 (8) | 101.466 | 101.758 | ||||
| C11-P2-C29 | 98.8 (2) | 97.683 | 97.896 | ||||
| P2-C11-P10 | 107.8 (8) | 116.659 | 117.127 | ||||
| C11-P10-C31 | 107.2 (2) | 106.537 | 106.503 | ||||
| C11-P10-C20 | 106.7 (2) | 105.185 | 105.532 | ||||
| C1-P10-C11 | 113.3 (2) | 116.705 | 116.610 | ||||
| C1-P10-C31 | 113.3 (2) | 104.958 | 105.045 | ||||
| P10-C1-H38 | 113 (3) | 117.095 | 116.835 | ||||
| C3-C1-P10 | 115.3 (3) | 116.926 | 117.816 | ||||
| C1-C3-O36 | 121.0 (4) | 121.327 | 121.402 | ||||
| C4-C3-O36 | 117.9 (4) | 118.453 | 118.528 | ||||
| C4-C9-H42 | 119.2 (5) | 117.402 | 117.777 | ||||
| C4-C5-H39 | 120.0 (5) | 120.260 | 120.230 | ||||
| C8-C7-Cl37 | 120.0 (4) | 119.650 | 119.575 | ||||
| C6-C7-Cl37 | 118.7 (4) | 119.525 | 119.442 |
Selected bond distances (Å) and angles (°) for complex C2.
| Bond distances | BP86/SVP | BP86/TZVP | Bond angles | BP86/SVP | BP86/TZVP |
| Pd10-C2 | 2.178 | 2.197 | C2-Pd1-P4 | 89.297 | 88.475 |
| Pd1-P3 | 2.422 | 2.422 | C2-Pd1-P3 | 90.334 | 91.296 |
| Pd1-P4 | 2.373 | 2.363 | C2-Pd1-P5 | 174.249 | 174.600 |
| Pd1-P5 | 2.401 | 2.379 | P3-Pd1-P5 | 83.960 | 83.611 |
| P3-C17 | 1.885 | 1.870 | P3-Pd1-P4 | 170.315 | 168.322 |
| P3-C18 | 1.838 | 1.828 | P4-Pd1-P5 | 96.449 | 96.876 |
| P3-C47 | 1.841 | 1.830 | Pd1-C2-C6 | 117.579 | 117.451 |
| P5-C16 | 1.869 | 1.856 | Pd1-C2-P14 | 117.536 | 117.694 |
| P5-C58 | 1.839 | 1.827 | Pd1-C2-H67 | 101.196 | 100.224 |
| P5-C63 | 1.840 | 1.829 | Pd1-P4-C15 | 106.967 | 107.406 |
| P4-C15 | 1.884 | 1.871 | Pd1-P4-C29 | 117.606 | 116.306 |
| P4-C29 | 1.846 | 1.835 | Pd1-P4-C41 | 119.085 | 119.959 |
| P4-C41 | 1.839 | 1.828 | Pd1-P3-C17 | 106.802 | 106.681 |
| C2-C6 | 1.536 | 1.532 | Pd1-P3-C18 | 113.699 | 112.977 |
| C2-P14 | 1.840 | 1.822 | Pd1-P3-C47 | 120.802 | 121.965 |
| C2-H67 | 1.111 | 1.103 | Pd1-P5-C16 | 105.434 | 105.449 |
| P14-C15 | 1.838 | 1.828 | Pd1-P5-C58 | 114.958 | 116.228 |
| P14-C32 | 1.827 | 1.816 | Pd1-P5-C63 | 121.344 | 120.729 |
| P14-C49 | 1.822 | 1.808 | P3-C17-C16 | 112.174 | 111.668 |
| C2-P14 | 1.840 | 1.822 | P5-C16-C17 | 109.265 | 108.765 |
| C15-H72 | 1.109 | 1.099 | C2-P14-C15 | 106.897 | 106.368 |
| C15-H73 | 1.109 | 1.100 | P4-C15-P14 | 112.278 | 112.623 |
| C6-O66 | 1.237 | 1.234 | C2-C6-O66 | 117.898 | 117.760 |
| C6-C7 | 1.490 | 1.486 | C2-C6-C7 | 121.260 | 121.516 |
| C16-C17 | 1.526 | 1.524 | C7-C6-O66 | 120.841 | 120.723 |
| C17-H76 | 1.110 | 1.100 | P14-C15-H72 | 111.966 | 112.210 |
| C17-H77 | 1.109 | 1.099 | P14-C15-H73 | 106.056 | 105.762 |
| C16-H74 | 1.109 | 1.099 | P3-C17-H76 | 105.834 | 106.259 |
| C16-H75 | 1.111 | 1.101 | P3-C17-H77 | 108.388 | 108.684 |
| C10-Cl13 | 1.737 | 1.732 |
4.10.2 1H NMR calculations
In order to correctly describe the experimental data, quantum chemical calculations of shielding constants were performed for the optimized structures of Y2, C2 and dppe. The absolute isotropic magnetic shielding constants (σi) were used to obtain the chemical shifts (Eq. (1)) by referring to the standard compound tetramethylsilane (TMS) for H atoms. The referred compound TMS were calculated at the same levels of theory.
| (1) |
The shielding constants are presented as chemical shifts and are compared with the corresponding experimental data (Table 7). The computed 1H chemical shifts of all remarkable protons in these compounds are in good agreement with the corresponding experimental values. As can be seen in Fig. 8, there is a good correlation between the experimental and calculated chemical shifts using both the TZVP and SVP basis sets. Thus it seems that the optimized gas phase structures are close to the structures of these compounds in solution.
The experimental and theoretical 1H NMR chemical shifts (δ, ppm) from TMS for the studied compounds.
| Compound | Exp | Calcda | Atom | Assignment | |
| BP86/SVP | BP86/TZVP | ||||
| Dppe | 2.1 | 1.7 | 1.7 | H29 | CH2 |
| 2.1 | 1.7 | 1.7 | H31 | ||
| 2.1 | 1.8 | 1.8 | H30 | ||
| 2.1 | 1.8 | 1.8 | H32 | ||
| Y2 | 3.6 | 3.0 | 3.1 | H43 | CH2 |
| 3.6 | 3.0 | 3.1 | H44 | ||
| 4.2 | 3.8 | 4.2 | H38 | CH | |
| 6.9 | 6.6 | 6.8 | H40 | Phenyl-Cl | |
| 6.9 | 6.8 | 7.1 | H41 | ||
| 7.1 | 6.9 | 7.1 | H39 | ||
| 7.1 | 6.9 | 8.1 | H42 | ||
| C2 | 2.66 | 1.9 | 1.5 | H76 | CH2 |
| 2.85 | 2.7 | 2.3 | H75 | ||
| 2.66 | 1.9 | 1.5 | H77 | ||
| 2.85 | 2.7 | 2.3 | H74 | ||
| 4.6 | 3.9 | 3.7 | H73 | CH2 | |
| 4.6 | 3.9 | 3.7 | H72 | ||
| 4.8 | 5.0 | 4.9 | H67 | CH | |
| 6.96 | 6.3 | 6.2 | H69 | Phenyl-Cl | |
| 6.98 | 6.6 | 6.4 | H70 | ||
| 6.99 | 6.8 | 6.6 | H71 | ||
| 7.01 | 7.2 | 6.9 | H68 |
a Using GIAO method.
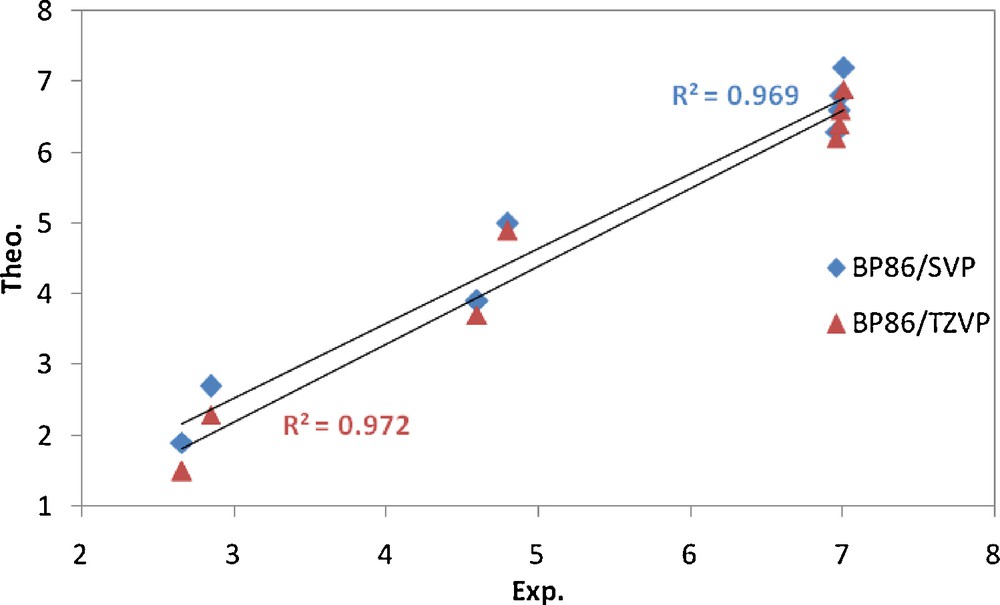
Correlation between theoretical and corresponding experimental values (δ, ppm) of 1H chemical shifts for complex C2.
4.10.3 31P NMR calculations
The absolute isotropic chemical shielding values (σiso) can be converted to chemical shifts (δ) relative to 85% aqueous phosphoric acid by comparison to the chemical shielding of PH3 at the same level of theory [93–96]. Thus we used Eq. (2) which is reported for calculation of phosphorus chemical shifts relative to 85% H3PO4 via:
| (2) |
Experimental data and theoretical phosphorus electronic shielding calculations of complex C2, phosphorus ylide Y2 and dppe ligand are presented in Table 8.
The experimental and theoretical 31P NMR chemical shifts δ (ppm) from H3PO4 for complex C2, phosphorus ylide Y2 and dppe ligand.
| Compound | Exp | Calcda | Atoms | Assignment | |
| BP86/SVP | BP86/TZVP | ||||
| dppe | −15 | −16.2 | 96.26 | P1 | |
| −15 | −16.2 | 96.26 | P4 | ||
| Y2 | 11 | −14.9 | 99.32 | P10 | |
| −30 | −31.5 | 69.89 | P2 | ||
| C2 | 37.4 | 15.0 | 11.32 | P14 | Pd |
| 19.5 | 24.0 | 17.32 | P4 | Pc | |
| 55.1 | 48.0 | 41.37 | P5 | Pa | |
| 54.9 | 55.0 | 48.25 | P3 | Pb |
a Using GIAO method.
It should be noted that the precise estimation of phosphorus NMR chemical shift is still an unsolved and very difficult problem [97]. However, the calculated GAIO 31P NMR chemical shifts of four types of phosphorus atom in complex C2 calculated at bp86/SVP level of theory are very close to the corresponding experimental values.
4.10.4 Interaction energies
The synthesized complexes have [(dppe)Pd(Y)](OTf)2 general formula. We were interested to study the interaction energies of ligands dppe and ylide Y with the Pd2+ metal ion. Three types of interaction energies in cation of the complex C2, [(dppe)Pd(Y2)]2+, were calculated with considering different fragments: (i) Pd2+ and both ligands dppe and Y, (ii) {(dppe)Pd}2+ fragment and Y, (iii) dppe and {Pd(Y)}2+ fragment. The following equations were used to calculate the interaction energies between the fragments in types i, ii and iii described above.
- (i)
- (ii)
- (iii)
In all cases, the charge of 2+ and a spin multiplicity of 1 were considered for the cation complex and also the fragments containing Pd ion. The calculated electronic energies of all species are given in the supplementary information (Table S1) and calculated interaction energies between the considered fragments are presented in Table 9.
Interaction energies in [(dppe)Pd(Y2)]2+ cation of complex C2 with considering different fragments.
| Method | ΔEint | ||||
| Pd2+: [(Y2)(dppe)] | [Pd(dppe)]2+: (Y2) | [Pd(Y2)]2+: (dppe) | Pd2+: (Y) | Pd2+: (dppe) | |
| BP86/SVP | −1070.531 | −139.307 | −122.364 | −980.797 | −946.284 |
| BP86/TZVP | −1039.155 | −131.149 | −119.854 | −949.421 | −922.438 |
As can be seen, the interaction energy between the Y2 and [Pd(dppe)]2+ fragment in the cation of complex C2 is more than that between dppe and [(Y2)(dppe)]2+. Furthermore, the sum of the above interaction energies is significantly less than that between the Pd2+ ion and {(dppe)(Y2)} fragment which includes both the dppe and Y2. Thus in order to understand the origin of this considerable difference, we calculated the interaction energy between the Pd2+ and each of the dppe and Y2 alone. As can be seen in Table 9, the interaction energy of the Pd2+ ion and Y2 is larger than that with the dppe, and both are significantly larger than those between the dppe and [(Y2)(dppe)]2+ and/or Y2 and [Pd(dppe)]2+ fragments. Thus the above data reveal three facts; (a) the bonding interaction of a Pd2+ ion with a single Y2 and/or dppe is very large; (b) the bonding interaction of Pd2+ ion with the Y2 is stronger than that between the Pd2+ and dppe; (c) the first coordinated bidentate ligand decreases significantly the interaction energy of the metal ion (the resulting fragment) with the second bidentate ligand.
4.11 Biological activity
4.11.1 DPPH radical scavenging assay
DPPH assay provided information on the reactivity of the complexes with a stable free radical. Because of the odd electron, DPPH shows a strong absorption band at 517 nm in visible spectrophotometery. As this electron becomes paired off in the presence of free radical scavenger, the absorption vanishes and the resulting decolorization is stoichiometric with respect to the number of electrons taken up [48]. Antioxidant properties, especially radical scavenging activities, are very important due to the deleterious role of free radicals in foods and biological systems [98]. Assessment of antioxidant activities showed that activity of the complexes in scavenging of free radical DPPH is fairly good but less than ascorbic acid (79.61%) as positive control. However there are no significant differences between scavenging ability of the free radical DPPH of complexes (Table 10). Antioxidant activities of the complexes did not promoted by increasing concentration.
Comparison of DPPH radical scavenging abilities of the studied complexes and ascorbic acid.
| Compounds | Concentration mg/ml (μM/l) | DPPH scavenging (%) |
| C1 | 0.2 (92.75) | 28.10 ± 2.8a |
| 0.4 (123.6) | 28.16 ± 2.6a | |
| 0.6 (185.5) | 29.55 ± 3.2a | |
| 0.8 (371.0) | 31.14 ± 1.8ab | |
| 1 (742.0) | 35.80 ± 2.5b | |
| Average | 30.55a | |
| C2 | 0.2 (93.25) | 30.11 ± 2.5a |
| 0.4 (124.3) | 31.85 ± 3.4a | |
| 0.6 (186.5) | 32.50 ± 1.8a | |
| 0.8 (373.0) | 32.38 ± 2.0a | |
| 1 (746.0) | 37.46 ± 2.7b | |
| Average | 32.86a | |
| C3 | 0.2 (92.5) | 34.08 ± 2.0a |
| 0.4 (123.3) | 34.45 ± 2.4a | |
| 0.6 (185.0) | 35.10 ± 1.4a | |
| 0.8 (370.0) | 35.80 ± 1.9a | |
| 1 (740.0) | 38.20 ± 2.6a | |
| Average | 35.53a | |
| C4 | 0.2 (93.6) | 29.95 ± 2.9a |
| 0.4 (124.8) | 30.15 ± 1.7a | |
| 0.6 (187.25) | 30.25 ± 2.5a | |
| 0.8 (374.0) | 33.20 ± 1.5a | |
| 1 (749.0) | 34.80 ± 2.1a | |
| Average | 31.67a | |
| Ascorbic acid | 0.2 (709.75) | 75.00 ± 0.8a |
| 0.4 (946.3) | 80.84 ± 1.5a | |
| 0.6 (1419.5) | 81.21 ± 1.4b | |
| 0.8 (2839.0) | 80.84b ± 2.2b | |
| 1 (5678.0) | 80.16b ± 1.9b | |
| Average | 79.61b |
4.11.2 Antibacterial activity
All complexes showed antibacterial activities against bacteria tested especially on Gram positive ones. In contrast, Serratia marcescens (-) was the most resistant bacterium (Table 11). When antimicrobial activities of the tested samples were compared with those of reference antibiotics, the inhibitory potency of the tested complexes was found to be remarkable (Fig. 9). Although all bacteria tested are resistant to Penicillin, the antibacterial effects of all complexes were higher than those of Penicillin on these bacteria. As it is well known, S. aureus and Bacillus species, especially B. cereus, are agents of food poisoning [99].
Antibacterial activities of the studied complexes at different concentrations mg/ml (μM/l).
| Microorganism | Inhibition zone diameter (mm) | |||||||||||||
| C1 | C2 | C3 | C4 | Solution | Antibiotic | |||||||||
| 1 (742) | 0.1 (74.2) | 0.01 (7.42) | 1 (746) | 0.1 (74.6) | 0.01 (7.46) | 1 (740) | 0.1 (74.0) | 0.01 (7.40) | 1 (749) | 0.1 (74.9) | 0.01 (7.49) | Hexane | Gentamycin | |
| P. vulgaris (−) | 13a | 11b | 9c | 15a | 11b | 8c | 16a | 14b | 10c | 14a | 12b | 8c | 7 | 30 |
| B. cereus (+) | 11a | na | na | 10a | 8b | na | 8a | na | na | 9a | 8b | na | 8 | 25 |
| S. aureus (+) | 18a | 11b | na | 16a | 12b | 10c | 20a | 15b | 11c | 15a | 11b | 10c | 9 | 35 |
| S. marcescens (−) | na | na | na | na | na | na | na | na | na | na | na | na | na | 27 |
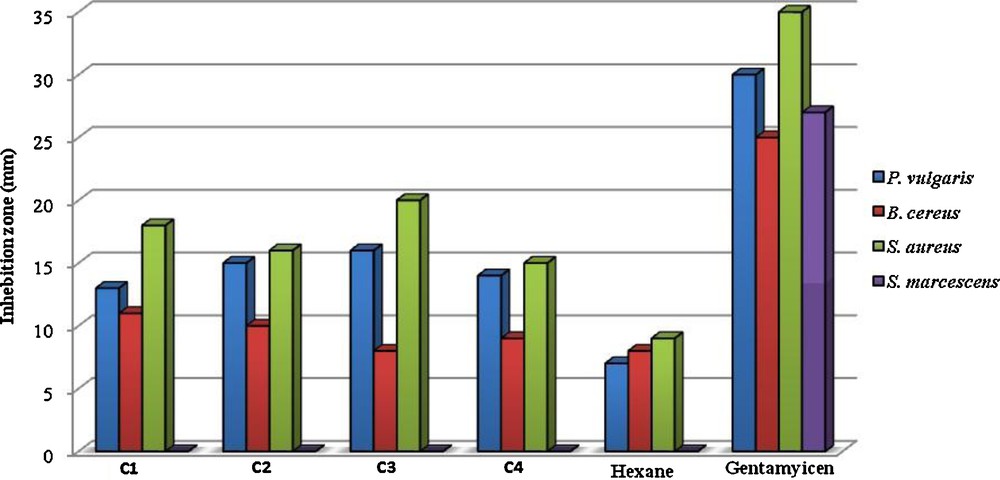
Antibacterial activities of the studied samples (at 1 mg/ml concentration), negative control (Hexane), and positive control (GM).
However, today it is known that the synergistic or antagonistic effect of one compound in minor percentage of mixture has to be considered. These complexes showed more activity against some bacteria under indicial experimental conditions. This would suggest that the structure of complexes may reduce the polarity of the metal ion mainly. Also, we can consider that the coordination may facilitate the ability of a complex to cross the lipid layer of the bacterial cell membrane and in this way may be affect the mechanisms of growth and development of microorganisms [100,101]. As can be seen in Table 11, the presence of Br, Cl, NO2 and OCH3 groups exerts a number of changes on antibacterial activities of the tested complexes. So, in case of C3, the presence of NO2 moiety induced an increase of their action against most of bacteria, while complex C4 was found to have low activity against bacteria. The above results indicate that the complexes studied may be used in treatment of diseases caused by bacteria tested. However, further studies are needed to evaluate the in vivo potential of these compounds in animal models.
5 Conclusion
The present study describes the very simple and efficient synthesis and characterization of a series of chelate Pd (II) complexes. The coordination ability of the phosphorus ylides and dppe has been proved in the complexation reaction with Pd (II) ion. Based on the results obtained from the physicochemical techniques of the metal complexes, one can conclude that the studied ligands behaves as a neutral bidentate ligand coordinated to the metal ion via phosphorus atoms of dppe in one side, and via terminal phosphorus atom (Pc) and methene group (CH) of the ylide on the other side. Thus, distorted square planar geometry is suggested for the studied complexes. In these distorted square planar complexes the three P-substituted atoms are coordinated; the fourth coordination site is filled with a methene group. Theoretical studies confirm a distorted square planner structure for the complex cation in C2 and probably other complexes. There is an excellent correlation between the calculated and experimental 1H NMR chemical shifts for latter complex. Calculated data also show that the bonding interaction of Pd2+ ion with the Y2 is stronger than that between the Pd2+ and dppe. Results from present study clearly demonstrated that the complexes exhibit antioxidant and antibacterial properties that might be helpful in preventing the progress of various diseases and can be used in an alternative system of medicine. However, possible side deleterious effects of these complexes on human health should be more investigated.
Acknowledgment
This work was supported by Bu-Ali Sina University funds. We thank Mrs. Akhlaghi for recording ESI-MS spectra. Our gratitude also goes to Mr. Zebarjadian for recording NMR spectra.