

1 General introduction
Polymerisation chemistry is one of the cornerstones of the modern chemical industry and has been at the forefront of both industrial and academic research and development for well-over 100 years [1–3]. Despite the monumental advances in polymer science, there is still a great need for novel polymer architectures and new polymerisation protocols. This requirement has been fuelled by such facets as the desire to increase energy efficiency in polymer production, reduce waste (i.e., “Greener” chemical technologies) and provide higher flexibility in the properties of the resulting polymers. This latter aspect has obvious relevance to new applications such as those in materials science and nanotechnology [1–6]. Traditional free-radical initiated polymerisation has been partially circumvented over time to the use of both cationic and anionic-mediated processes in an attempt to exhibit more precise control over polymer molecular weights, dispersities1 and ultimately bulk macro-scale polymer properties [1–3]. Controlled radical polymerisation, involving stable radicals such as 2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (tempo) or paramagnetic transition metal species [8,9], has recently been at the forefront of new synthetic methods which display greater control of the growth of the polymer chain(s), in addition to the usefulness of this technology in block co-polymer syntheses. Hence, metal-mediated polymerisation offers a well-established alternative to traditional free-radical chain growth methods and is a very useful supplement to the variety of anionic and cationic-polymerisation mechanisms [10] that are currently known. Some thirty years ago, it was realised that metallocene-based (i.e., cyclopentadienyl: Cp) systems, specifically those incorporating Ti or Zr, could be fashioned to produce highly active polymerisation catalysts that engender facile control of polymer molecular weights and dispersities. This resulted in somewhat of a renaissance within organometallic chemistry and led to intense research efforts in this area; these investigations have culminated with the widespread industrial application of single-site metallocenes for polymer production [6,8–13]. The later introduction of chiral versions of these catalysts, specifically the ansa-metallocenes [1–3,5,13], allows for the selective production of stereo-regular linear polymers with rich enhancement of either iso-tactic or syndio-tactic architectures (Fig. 1). Most of these systems require alkyl-aluminum compounds, or the polymeric version Methylalumoxane (mao), in large stoichiometric excess (> 500 equivalents) to operate efficiently. These latter “co-catalysts” or “activators” likely serve to rigorously dry the reaction medium while concomitantly generating the active alkyl-M (M = Ti, Zr, etc.) polymerisation catalyst. Not surprisingly, the early transition metal pre-catalysts can be expensive to produce and are often highly sensitive to the presence of traces of water.
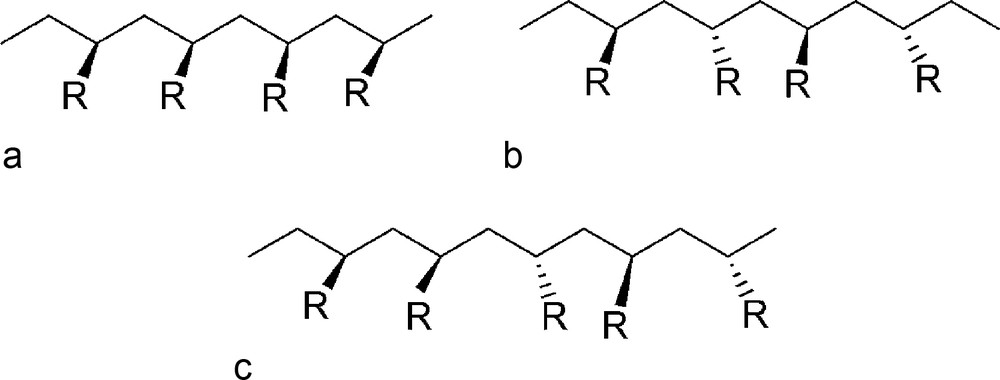
Schematic representations of atactic and stereo-regular polymers (R can be any functional group except H). a: iso-tactic: the configuration at the stereogenic site in each repeating is identical; b: syndio-tactic: the configuration at the stereogenic site in each repeating alternates; c: atactic configuration: the configuration at the stereogenic site in each repeating is random.
Historically, tight patent control over many of the Ti and Zr systems spurned significant research and development and this has resulted in the introduction of a wide variety of non-metallocene systems [14] that impart similar activities in monomer activation. In 1995, Brookhart and co-workers disclosed [15,16] that late transition metal complexes of α-diimines, specifically that of Pd and Ni, could act as highly active catalysts for the polymerisation of olefins such as ethylene [15–25]. This area has expanded in recent years into a variety of homo- and co-polymerisation processes which have been the subject of several recent reviews [14,16,18,19,21,22]. Stereo-specific polymerisation (i.e., iso- or syndio-tactic product enhancement) by late transition metal systems, especially Ni, is however much less explored [25,27] and is still a subject of significant research endeavours. In this brief Account, we bring forth a few recent examples of Ni-based systems intended for the production of polymers (e.g., polyethylene) including those of enhanced stereo-regularity which incorporate monomers such as styrene and more polar monomers such as methyl methacrylate.
2 Mononuclear nickel systems for stereo-regular polymer syntheses
It has been known for many years that π-allyl complexes of Ni can convert monomers such as butadiene into high polymers. However, the rates of polymer formation and yields of the resulting polymer materials are typically similar to that displayed by the more traditional Ziegler-Natta type systems [1]. Later work by Shell Chemicals introduced the “shop” (Shell Higher Olefin Process) catalysts [1–3,25] consisting of mononuclear formal Ni2+ metal centres and chelating P,O-donor ligands (Fig. 2: 1). Although these species demonstrate good activity in oligomerisation chemistry, they are relatively sluggish in terms of general applicability for bulk polymerisation. Despite these drawbacks, ligand design strategies based on the shop-type systems continue to be an area of active research [1–3,5,28,29]. The introduction of the Brookhart-type catalysts, specifically Ni(α-diimine)X2 species 2 and 3 (Fig. 2) in 1995, greatly changed the landscape of Group 10-based catalysts systems. These species, and in particular the cations derived from them via halide abstraction, are highly active catalysts for polymerisation of apolar monomers such as ethylene and propylene [15,16]. In the ensuing years, a very large number of papers and patents have been disclosed detailing modifications and improvements to the α-diimine-derived catalysts [28,29].
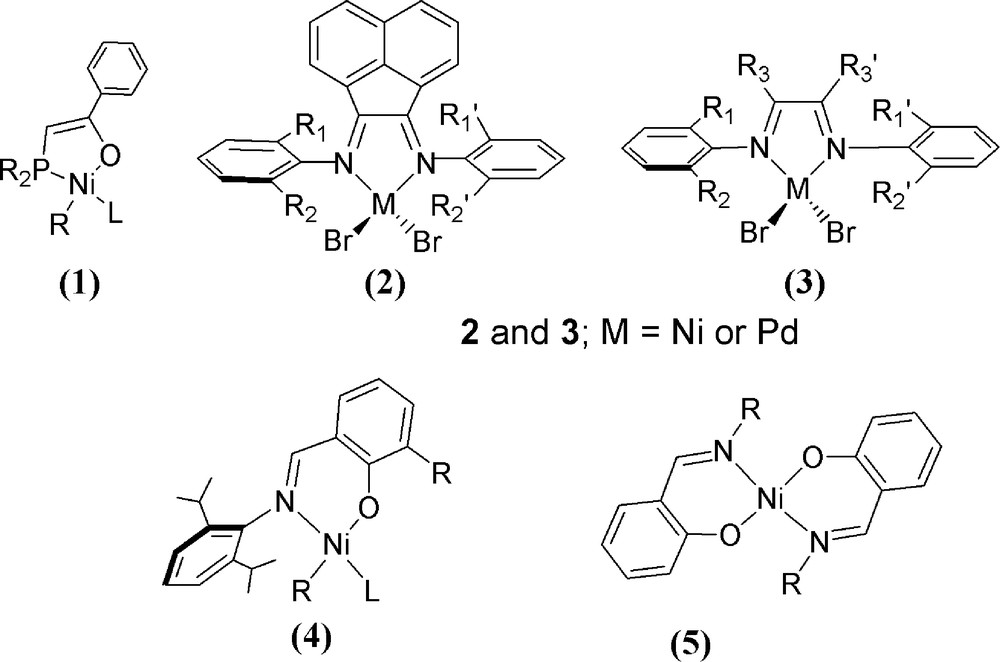
Schematic representations of the “shop” catalysts (1), the Ni/Pd-based “Brookhart”-type catalysts (2 and 3), the Grubbs’ bis-iminophenolato polymerisation catalysts (4) and a generic bis-imine Ni pre-catalyst (5).
Some five years later, Grubbs disclosed a series of aryl-Ni κ2-N,O-iminophenolato (4: Fig. 2) compounds [25,30] that are effective for both homo- and co-polymerisation of ethylene and polar monomers; the latter of which can contain sensitive functional groups and heteroatom centres. These systems can also operate in polar solvents [25,28,30]. Extensions into more air-stable compounds have included a large number of studies of Ni bis-iminophenolatos (Fig. 2: 5) which generally display useful ethylene and norbornene polymerisation profiles [28,31]. Nickel complexes have also been employed for controlled radical polymerisation; these catalysts include examples of Ni-pincers and Ni phosphine complexes although their activities are generally lower than that observed for other transition metals [8c]. Much of the chemistry discussed in this section thus far is certainly seminal, but much less work has been directed towards stereo-specific polymerisation. Early work with simple systems such as Ni(acac)2 in the presence of mao revealed that typically a random polymer, such as that of atactic-polystyrene, was produced with somewhat disappointing reaction kinetics [32]. Attempts to modify this system by employing other Ni derivatives and or additional ligands, solid supports or alternative alkyl-aluminum sources has led to moderate successes in the formation of iso-enhanced (Fig. 1) polystyrene [33].
The Ni-indenyl salt complex 6 (Fig. 3), reported by Groux and Zargarian [34], displays good polymerisation activity with both styrene and/or norbornene (80 °C) to give moderate molecular weight polymers with good dispersity values. However, no stereo-specificity for polymerisation with 6 was noted at the time. Studies on the closely related three indenyl complexes 7 (Fig. 3) also demonstrate good styrene polymerisation activities upon activation with NaBPh4 with or without the addition of excess PPh3. In this latter case however, syndio-rich atactic polystyrene can be obtained [35].
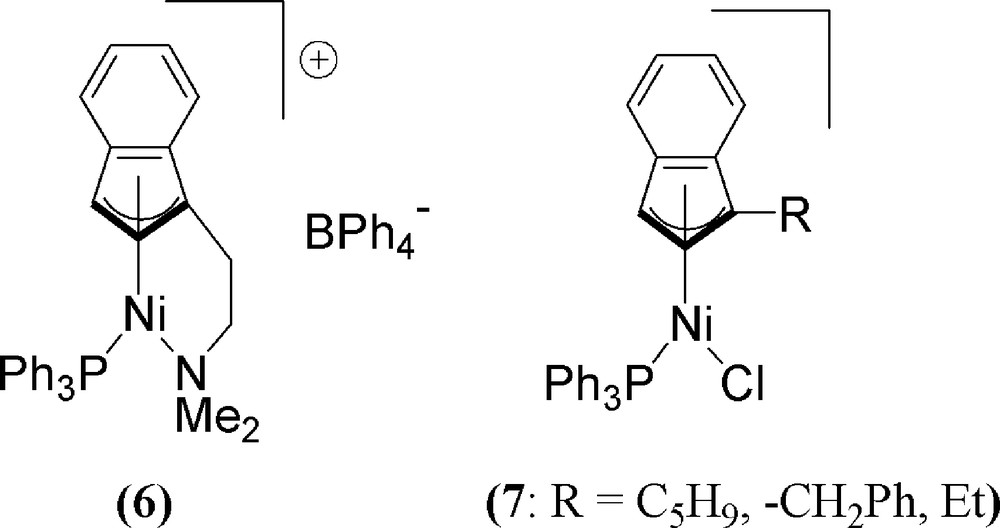
The aminoindenyl Ni polymerisation pre-catalyst of Groux and Zargarian (6) and related derivatives 7 demonstrating syndio-rich styrene polymerisation.
The potent nature of the Brookhart class of catalysts has led to, perhaps not surprisingly, extensive modification of the α-diimine skeleton (e.g., Fig. 2: 2 and 3). These endeavours are focused on the dual objectives of increasing reaction kinetics while also further controlling the resulting polymer architectures for monomers beyond that of ethylene [4,5,8c,9,12,14,17–22,26,28]. Early success with a modified bis-imine system by Pellecchia and Zambelli revealed that syndio-tactic specific polymerisation could be obtained for poly-propylene at low temperatures [36]. The more constrained ligand designs revealed by Coates et al. [37] (2: Fig. 2) give iso-rich polymers in combination with attractive kinetics and the capability to form block co-polymers which include long chain linear α-olefins such as 1-hexene or 1-octene. Recent work by the Bazan group has clearly demonstrated how subtle changes in ligand design, such as the introduction of a keto group on the ligand backbone (8: Fig. 4), can dramatically improve catalyst performance, enable block co-polymer formation and provide enantio-control (iso-tactic) of the resulting polymer skeletons [38].
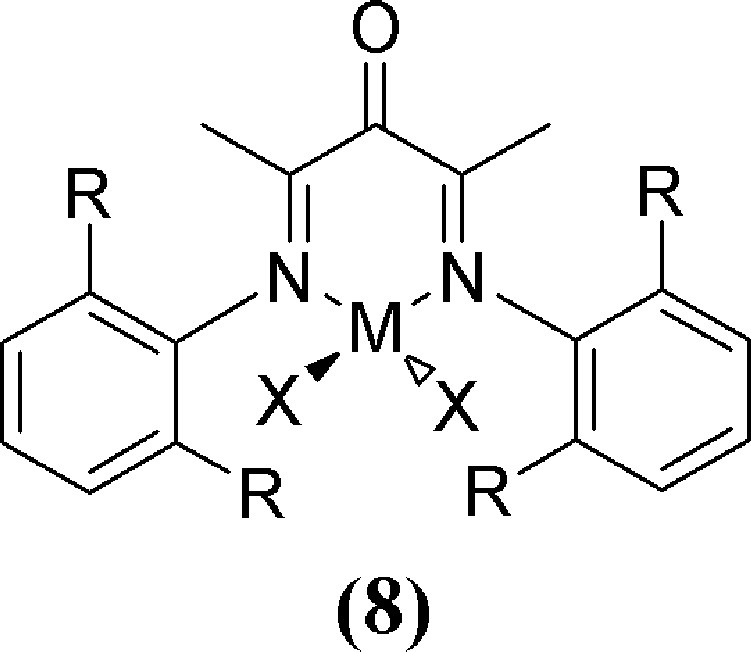
Bazan's α-keto-β-diimine Ni design (8) for iso-selective olefin polymerisation.
3 Di- and multi-nuclear nickel complexes in polymer syntheses
The successful implementation of mononuclear Ni-based catalysts for polymerisation over the last 20 years has in recent times expanded to include di- and multi-nuclear Ni species. Many of these systems show intriguing cooperative effects on the catalytic profile although the design and synthesis of ligand frameworks that can accommodate two or more metal centres is obviously more involved when compared to that of single-site catalysts. A number of these designs are similar in style to those schematically depicted in Fig. 5 as pictorial compounds 9 and 10. Hence, bridge design strategies have enabled the incorporation of two Ni atoms in (typically) relatively close proximity to one another. The catalytic profiles of these species are then compared and contrasted to mononuclear model compounds. This area of research is still in its infancy but will no doubt continue to expand in the upcoming years. Readers are directed to the excellent 2011 review [28] by Delferro and Marks that encompasses all multi-nuclear polymerisation catalysts [28,39]. Therefore, we shall only briefly survey herein a few more recent examples.
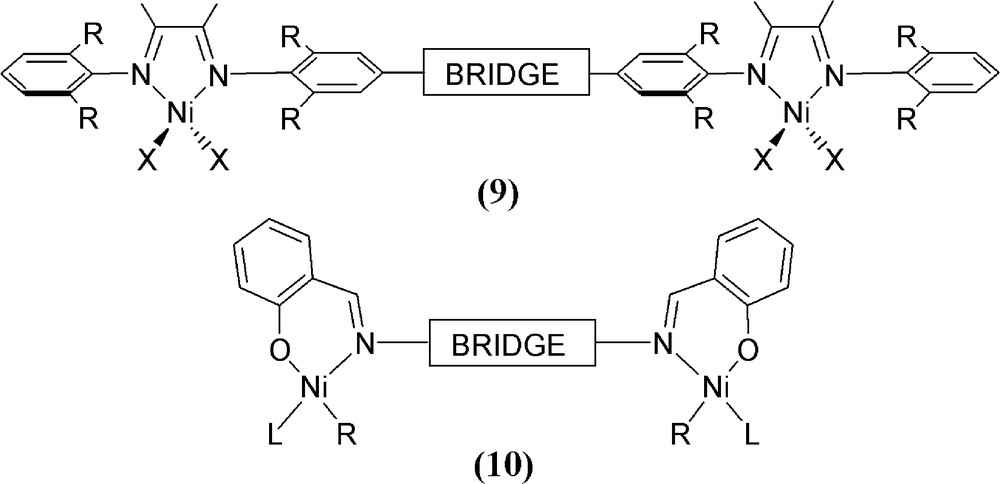
Some typical design strategies for di-nuclear Ni polymerisation catalysts.
Recent works by Marks and co-workers on napthoxyiminato systems include a detailed study of di-nuclear complexes such as 11 (Fig. 6). These materials show increased stability and potent ethylene polymerisation capability (with co-catalysts) when compared to their mononuclear analogues. In addition, there is an increased propensity to give more branched polyethylene products and a dramatic increase in norbornene incorporation in ethylene-norbornene co-polymerisation reactions [40]. Agapie's bis-nickel phenoxyiminato compounds (Fig. 6: 12) can exist in two isomeric forms (syn- or anti-) and are active for polymerisation of C2H4 and ethylene/α-olefin co-polymerisation. The two isomers demonstrate differing levels of activity with the anti-isomer being more active [41]. Halogenido-bridged dimeric catalysts 13 (Fig. 5) are highly active catalysts in the presence of excess mao for the production of polyethylene in which low Đ values are obtained with a high degree of branching [42]. Also in this case, the di-nuclear materials are more active than their mononuclear congeners.
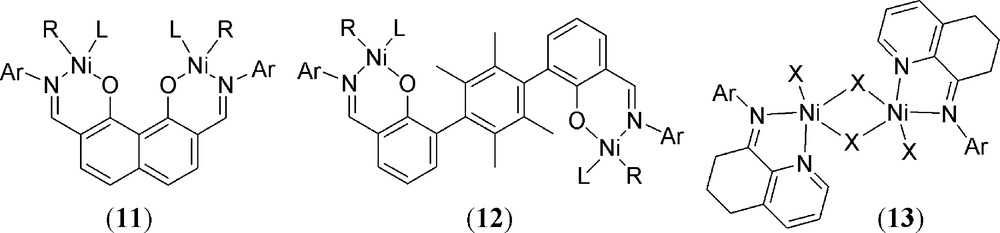
Recently tested bis-nickel polymerisation catalysts; 11: R = –CH3, L = PMe3, Ar = –C6H3-[CF3]2-2,6 or –C6H3-[CH3]2-2,6; 12: R = –CH3, L = pyridine, Ar = –C6H3-[i-Pr]2-2,6; 13: X = Cl or Br; Ar = –C6H2-[CHPh2]-2-R-4-R’-6; R and/or R’ = Me, i-Pr or –CHPh2.
Complexes such as 13, which contain both terminal and bridging halogenido ligands, presumably form catalytically active complexes due to interaction with the large excess of mao that is present. This strongly implies that one or more of the halogen atoms are replaced via alkylation by mao to create the active catalyst(s). The form and nature of these materials is often still the subject of debate as large amounts of alkyl-Al reagent are present, often several hundred fold in excess of the catalyst precursor. Indeed simple Ni complexes, such as the aforementioned Ni(acac)2 [32], do form active polymerisation catalysts with mao but the resulting polymer architectures are atactic in nature. In a similar way, a tri-nuclear α-diimine cluster reported by Helldörfer et al., 14 (Fig. 7: L = κ2-N,N’-Ar-N=C(Me)C(Me)=N-Ar; Ar = -C6H3-Me-2-Cl-4; X = E = Br) which contains Br atoms acting as bridging bromido, capping bromido and a non-coordinated bromide anion can be activated by excess mao to give a moderately active ethylene polymerisation catalyst [43]. In contrast, related clusters containing N,N,N’,N’-tetramethylethylenediamine (tmeda) as L (X = Cl; E = OH) form active catalysts with stoichiometric mao to yield syndio-rich polymers with styrene or methyl methacrylate as monomers [44]. Related Ni oxazoline-containing (i.e. imine, vide supra) clusters were shown to be primarily inactive under such conditions. The nature of the selectivity in polymer motif was probed using density functional theory (dft). In the case of 14 in which L = κ2-N,N’-tmeda and X = Cl; E = OH, the orientation of the diamine ligand backbones cause the three bridging chloride atoms to be chemically inequivalent [44]. This leads to a preference (ground state: dft) for chloride-methyl exchange at a bridging chlorido position in which the wide angle of the flanking ligand backbones (Fig. 8: halogen atom X in red) are oriented towards the predicted lumo in which an incoming olefin would attach (Fig. 9). Hence, a chiral pocket is created which gives a reasonable explanation for the syndio-rich nature of the resulting polymers [44]. It is certainly possible that reaction of complexes such as 14 with mao may lead to partial breakdown of such clusters to form achiral materials such as NiMe2(tmeda). This may help to explain the formation of some atactic units within the resulting polymers. Work on this latter system is still on-going [45].
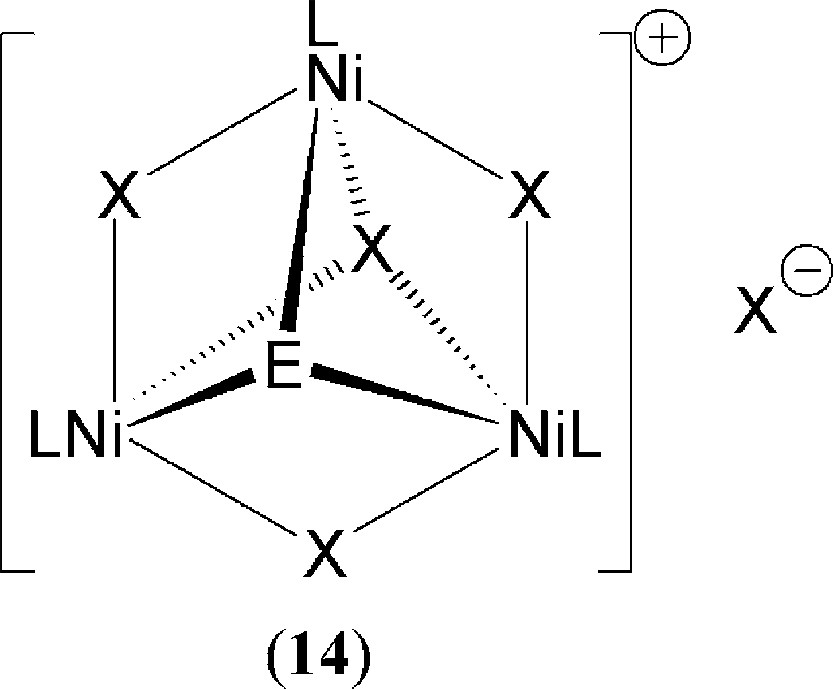
General schematic of known tri-nuclear clusters which are active polymerisation catalysts.
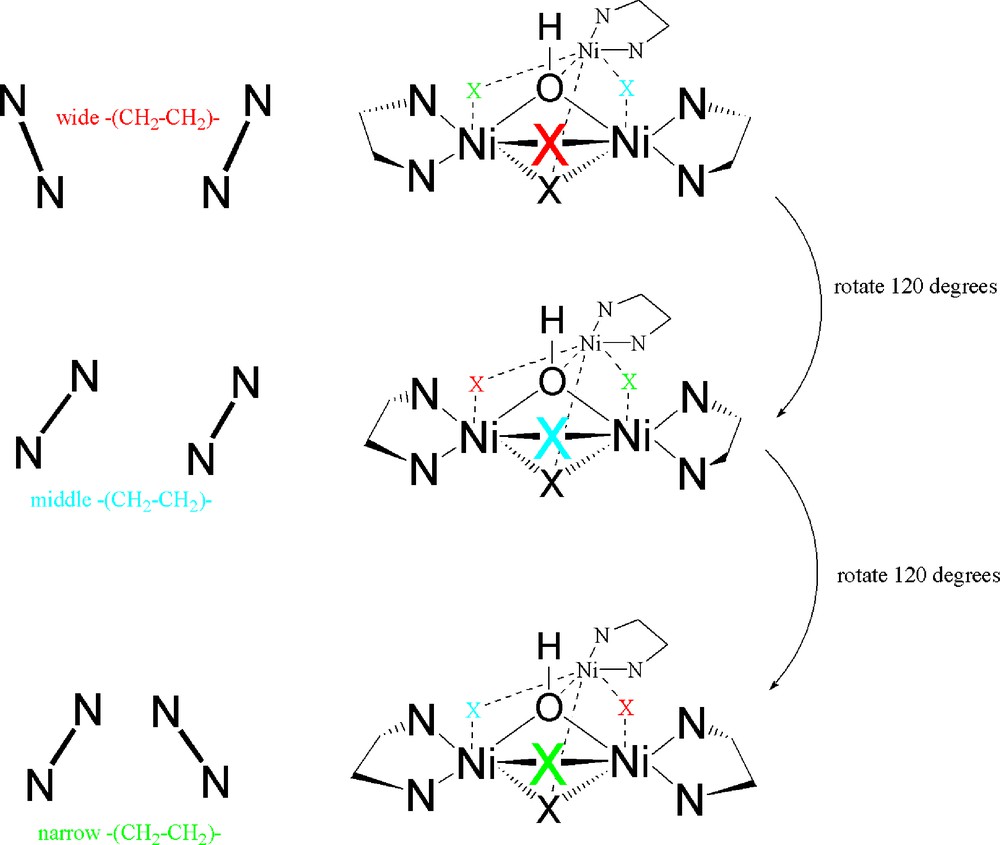
Schematic “side-on” view of a Ni3-cluster indicating the differentiation between bridging chlorido ligands.
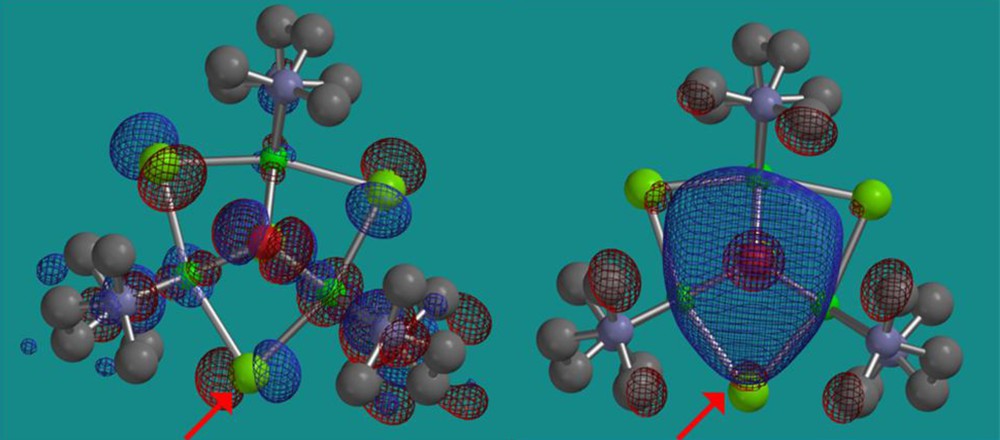
“Top-down” view of a tmeda Ni3-cluster looking down through the O-H bond showing the homo (left) and lumo (right); the energetically favourable Cl atom for replacement by CH3 is shown by the arrow (DFT: B3LYP/6-31G*/LANDL2DZ).
4 Concluding remarks
The renaissance of application in Ni-mediated catalysis, initiated by Brookhart's seminal work in the middle of the 1990s, continues to produce interesting and useful systems for not only homo-polymerisation but also for block co-polymer and stereo-specific polymerisation chemistry. The recent surge in di- and tri-nuclear nickel species with enhanced reactivities and selectivity suggest that a body of interesting work is just on the horizon in this area of catalysis.
Acknowledgements
The authors are indebted to the support of Ryerson University (Dean's Research Fund, Dean's Travel Fund), MaRS Innovation and NSERC (Canada) in the form of Discovery Grants (D.A.F., R.A.G.). Y.-Y.G.L. thanks Ryerson University for a graduate student scholarship. Dr. Catherine Bonnier (University of Toronto) is thanked for a critical review of this work. R.A.G. would also like to personally thank the committee of the gecom-concoord 2012 meeting (Métabief, France) for the invitation to submit this manuscript and for organizing such an excellent symposium. Several anonymous reviewers are also acknowledged for providing very helpful feedback and suggestions which greatly improved this manuscript.
1 The term “dispersity” (symbol Đ, i.e., “D-stroke”) has been recommended by IUPAC to replace terms such as “poly-dispersity index”, etc. and hence this term is used throughout this manuscript [7].