

1 Introduction
Stilbenoids are well-known compounds applied in the pharmaceutical industry. Some stilbenoids occur in nature, but their synthetic analogs are also known [1,2]. Naturally occurring stilbenoids are prominent building blocks in organic synthesis. Both (E)- and (Z)-stilbenoids have important biological activities [3,4]. They possess anti-oxidative, anti-microbial and anti-tumor properties [5], induce apoptosis (programmed cell death) [6], cause vascular disruption in tumors [7] or inhibit tubulin polymerization [8]. Among (E)-stilbenoids, resveratrol and its derivatives play a crucial role in cancer therapy due to their physiological and therapeutic properties [9]. Among (Z)-stilbenoids combretastatins possess the most interesting properties for pharmaceutical industry, due to their ability to inhibit tubulin polymerization [8].
For economic reasons, stable stilbenoids that can be isolated in large quantities from natural sources are most often examined for their utility as drugs components. For example, they can be isolated from grapes and combretastatins from the willow bush. Stilbenoids can also be modified to enhance their medicinal properties [10]. However, currently known synthetic methods for obtaining stilbenoids are frequently complex and not efficient enough. Therefore, it is important to find novel and simple methods to synthesize stilbenoid derivatives [11,12].
Stilbenes were initially obtained in Wittig [13] and Horner–Wadsworth–Emmons reactions [14]. Unfortunately, these reactions have led to the formation of carbon–carbon double bonds, providing unsatisfactory (E)/(Z) selectivity. To enhance the (E)/(Z) selectivity a number of transition metal-based catalysts were later used, such as palladium complexes in Heck [15], Negishi–Stille [16], Suzuki [17] and tandem [18] processes. Alternatively, low-valent titanium complexes were in a stoichiometric manner used for coupling two aldehydes or ketones in a McMurry reaction [19]. Moreover, in the presence of molybdenum [20] or ruthenium [21] complexes, a new carbon–carbon double bond can be directly formed in the olefin metathesis reaction between two styrenes. This method is very promising as it allows the use of a broad range of substrates and requires only a small amount of catalyst under relatively mild experimental conditions. However, problems with (E)/(Z) stereoselectivity and the tendency for the formation of the mixture of products due to cross-metathesis (CM) or homo-dimerization (HD) are the main drawbacks of this approach. Therefore, according to our knowledge, only a few examples of stilbene formation using alkene metathesis are reported in the literature [20–22].
In order to eliminate the drawbacks of the stilbenoid synthesis via metathesis, which are indicated above, we have investigated the system where two styrenes are connected by silicon linker [1,1,3,3-tetramethyldisiloxane], as shown in Fig. 1. Here, we describe the effect of the use of such linker in the stilbenoid synthesis on the selectivity of metathesis reaction using standard metathesis condition (dichloromethane [DCM] at 40 °C and toluene at 80 °C) and commercially available catalysts 1–3 (Fig. 2).
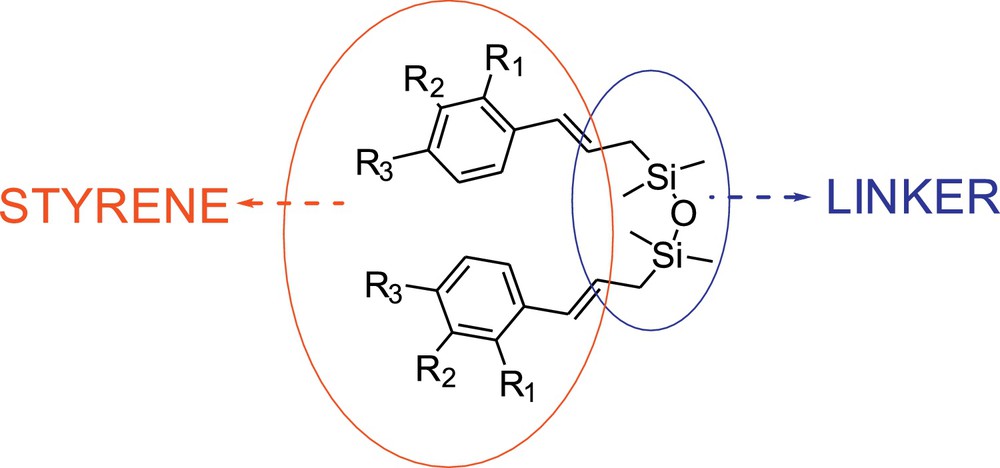
Substrates used in the present study.
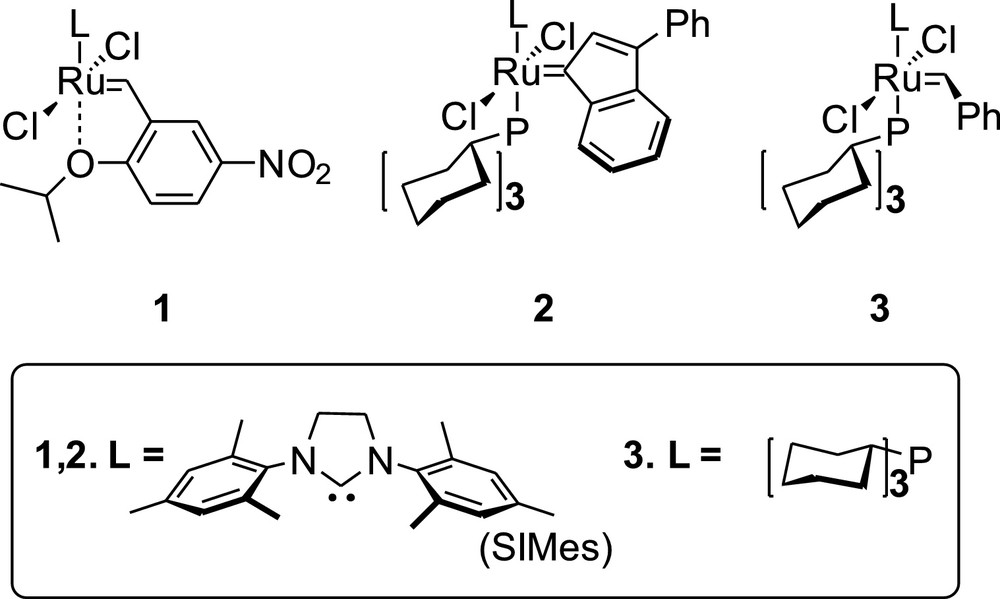
Structures of the ruthenium catalysts 1, 2 and 3 used in this study.
2 Results and discussion
2.1 Preparation of 1,3-dicinnamyl-1,1,3,3-tetramethyldisiloxane derivatives
For our studies, we have chosen 1,3-dicinnamyl-1,1,3,3-tetramethyldisiloxane derivatives (Fig. 3) as model compounds as in this case two styrenes are connected by silicon linker. Following the synthetic route presented in Fig. 3, desired lithium reagents were obtained by metalation of allyl-substituted arenes 4-7 by n-buthyllithium–tetramethylethylenediamine (n-BuLi–TMEDA) in tetrahydrofuran (THF) at 0 °C, resulting in the formation of lithium reagents 8–11, respectively. The completeness of the metalation step was guaranteed by rising the temperature from 0 to 25 °C. The reaction of cinammyllithium derivatives 8–11 with 0.6 equiv. of 1,3-dichloro-1,1,3,3-tetramethyldisiloxane afforded exclusively compounds 12–15. We obtained the products with (E)-isomer excess of 88–92%.
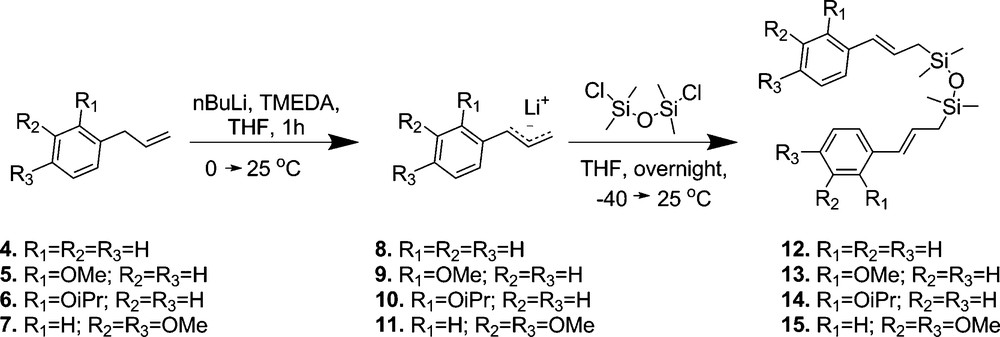
Synthetic route of 1,3-dicinnamyl-1,1,3,3-tetramethyldisiloxane derivatives.
Initially, we have examined the reactivity of compound 12 in olefin metathesis using complexes 1–3 (Fig. 1) at 40 °C in DCM and 80 °C in toluene (Table 1). The reaction did not proceed in the presence of the first-generation Grubbs catalyst 3 (5 mol%) in DCM at 40 °C (Table 1, entry 3), and 12 was still present in the reaction mixture, indicating no degradation of the substrate under the reaction conditions. Even an increase of temperature had no effect on the reaction performed with catalyst 3 (5 mol/%) in toluene at 80 °C (Table 1, entry 5). The use of catalysts 1 and 2 at 40 °C (Table 1, entry 1 and 2) led to the formation of stilbene in 44% and 91% yields, respectively. At high temperature (80 °C), catalyst 2 promoted the reaction very effectively, resulting in 90% of yield (Table 1, entry 4). In all cases the formation of stilbene has been proven by GC–MS analysis (Tab. I). In the case of reactions catalyzed by 1 and 2, we compared the retention times of the reaction product and the (Z)- and (E)-stilbene standards, and found that only (E)-isomer was formed.
Metathesis of substrate 12 witch catalysts 1–3.
| Catalyst | Solvent | Temperature (°C) | Yeld (%)a | |
| 1 | 1 | DCM | 40 | 44 |
| 2 | 2 | DCM | 40 | 91 |
| 3 | 3 | DCM | 40 | 0 |
| 4 | 2 | Toluene | 80 | 90 |
| 5 | 3 | Toluene | 80 | 0 |
a Yields determined by GC–MS analysis of the reaction mixture.
On the basis of initial results presented above, catalyst 2 (Umicore™ M2) was chosen for further examination of the metathesis reactions of compounds 13–15 (Figs. 4 and 5). The reactivity of substrate 15 was found to be similar to the reactivity of 12. Despite noticeable (E)/(Z) isomerisation of substrate 15 resulting in (E)/(Z) of 88/12, we observed that metathesis of 15 led exclusively to the (E)-stilbene and was also fast and efficient, and no other products were formed. However, both 13 and 14 were inactive towards metathesis and the formation of stilbene 17 and 18, respectively, was not observed (Table 1).

Reaction profiles of the compounds 12–15 with catalyst 2 (5 mol/%).
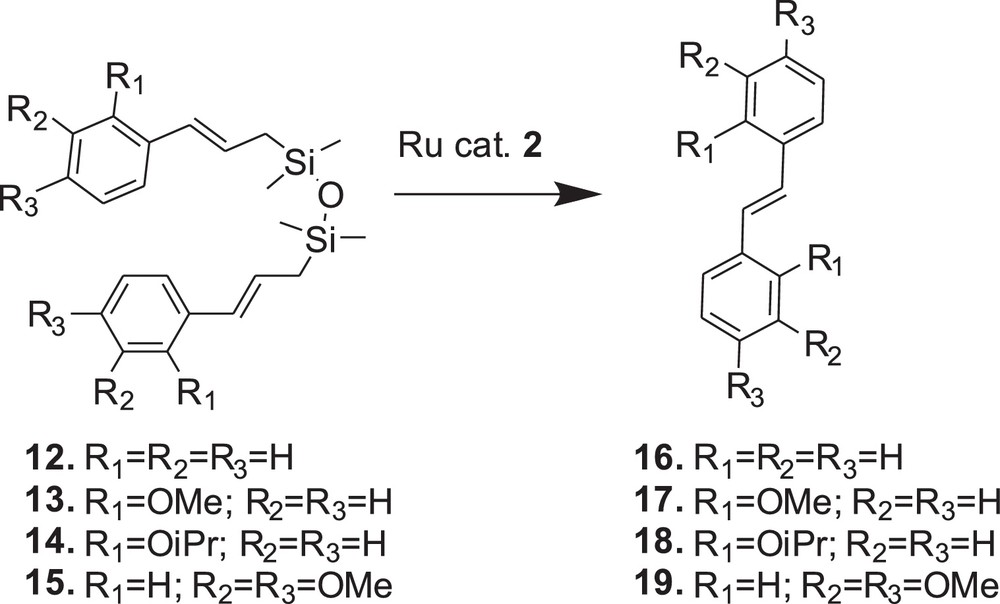
Metathesis reaction promoted by catalyst 2.
In order to explain the lack of metathesis activity of 13 and 14 catalyzed by 2, we performed the equimolar reaction between 13 and catalyst 2, obtaining compound 20 (Fig. 6). Compound 20 was isolated as green crystals in 49% yield. Noteworthy, the formation of the new catalyst inactive in the metathesis of 13, explains the lack of formation of 17. It should be noted that complex 20 [23] is a methyl analog of Hoveyda–Grubbs catalyst (21), which apparently exhibits low activity in olefin metathesis with this specific substrate. As 21 is expected to be formed in the case of an equimolar reaction between 14 and 2, the lack of formation of 18 should be explained by low catalytic activity of 21 in the metathesis of 14. In order to check the stability of substrates under the reaction conditions, we performed the stability tests in the absence of Ru catalysts by heating compounds 12–15 at 80 °C for 3 h. It was confirmed that the substrates are stable under these conditions. To support our findings discussed above, the formation of the catalyst 20 in the RCM of 13 prevents further metathesis reaction, we performed RCM of 13 with 1. For the latter the lack of RCM of 13 is in line with our earlier observations.

Formation of Hoveyda–Grubbs catalysts in reaction of 13 and 14.
In the next step, we performed the metathesis reaction using the mixture of compounds 12 and 15 in order to show whether inter- or intramolecular process operate in that case (Fig. 7). The metathesis of 12:15 (1:1) was catalyzed by 2 (5 mol%, c = 0.02 mol/L). We monitored the reaction progress by the GC–MS and observed that initially (after 15 min), only compound 23 was formed. After prolonged reaction time, the formation of compounds 16, 19 and 22 was observed. After 5 h, the products 16, 19 and 22 were present in 26:17:49 molar ratios, respectively. Noteworthy, the metathesis reaction of 12 + 15 (mixed in molar ratio 1:1) proceeded more slowly in comparison with the metathesis reaction performed separately for compound 12 or 15. This experiment indicates that both CM and RCM may operate in the investigated system. The control CM reaction with styrene and 3,4 dimethoxystyrene promoted with catalyst 2 (the experiment was performed analogously to the one with 12 and 15) shows that simple CM of these substrates leads to the formation of 16, 19 and 22 in 32:26:48 molar ratios, respectively. It shows that the presence of siloxane linker influences the distribution of products.
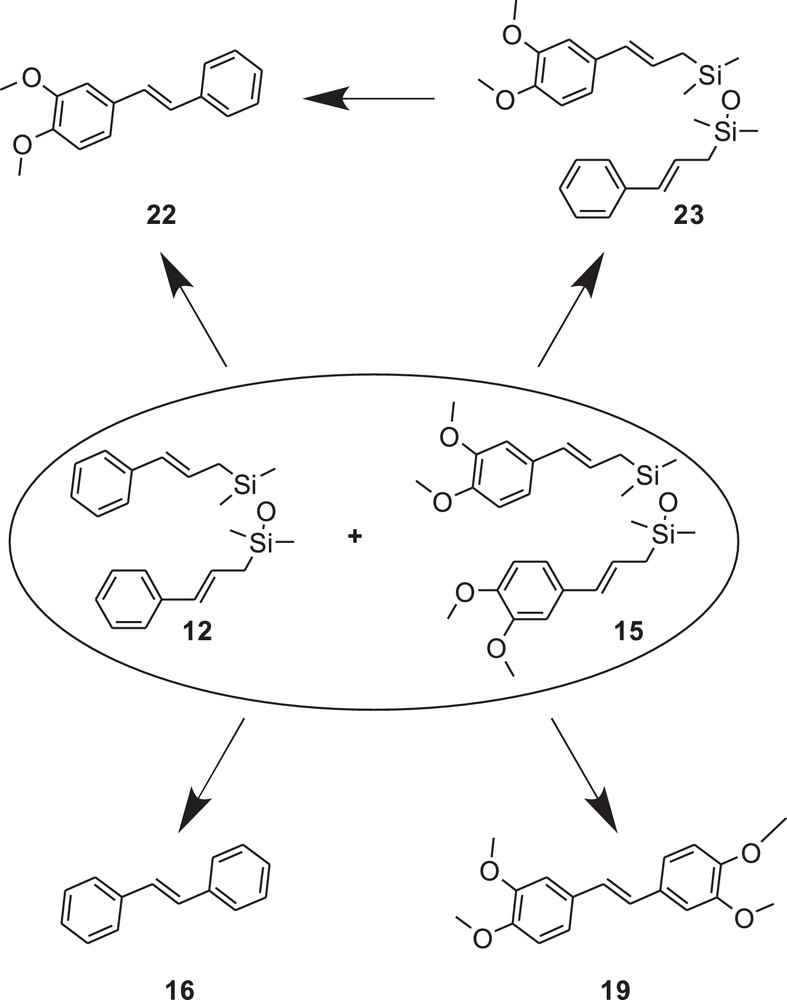
Structures of the possible products formed in the metathesis of 12+15 mixture.
In order to investigate the concentration effect on the product distribution, we also performed the experiment described above using two different concentrations, 0.1 mol/L or 0.02 mol/L, of both 12 and 15. After 2 h for the reaction in concentration 0.1 mol/L, we obtained stilbene derivatives 16,19 and 22 in 12:17:30 molar ratio, respectively. In the reaction mixture, a small amount of 23 was detected by GC–MS. For the concentration of 0.02 mol/L, we observed the same stilbene derivatives, in 14:16:35 molar ratio, respectively. The above study showed that concentration of 12 and 15 influences the composition of the products formed only slightly (Table 2).
Products formed in the metathesis of 12+15 mixture.
| Time (min) | Concentration (mol/L) | Yield (%)a | ||
| 16 | 19 | 22 | ||
| 5 | 0.1 | 0 | 0 | 0 |
| 0.02 | 0 | 0 | 0 | |
| 15 | 0.1 | 0 | 0 | 0 |
| 0.02 | 0 | 3 | 0 | |
| 30 | 0.1 | 0 | 5 | 7 |
| 0.02 | 1 | 4 | 4 | |
| 60 | 0.1 | 6 | 12 | 23 |
| 0.02 | 6 | 11 | 17 | |
| 120 | 0.1 | 11 | 16 | 30 |
| 0.02 | 14 | 16 | 35 | |
| 300 | 0.02 | 26 | 17 | 49 |
a Yields determined by GC–MS analysis of the reaction mixture.
After characterization of the styrene derivatives formed in metathesis of 12–15, we have focused on the fate of the disiloxane part. The GC–MS analysis of the reaction mixtures revealed the presence of only one silane-containing product for both 12 and 15. The GC–MS peak, representing this product had a retention time of 7.67 min. This retention time and the mass spectrum shown on (Fig. 9) indicate the presence of divinyldisiloxane 24, according to the mass spectra library. However, unambiguous identification was not possible due to some differences between the spectrum shown in Fig. 9 and the mass spectrum of compound 24 in the NIST library. Fragmentation process of both compounds (showed in Supplementary data, Figs. S3 and S4), most likely begins with the elimination of methyl radical leading to the formation of the ion m/z = 171.
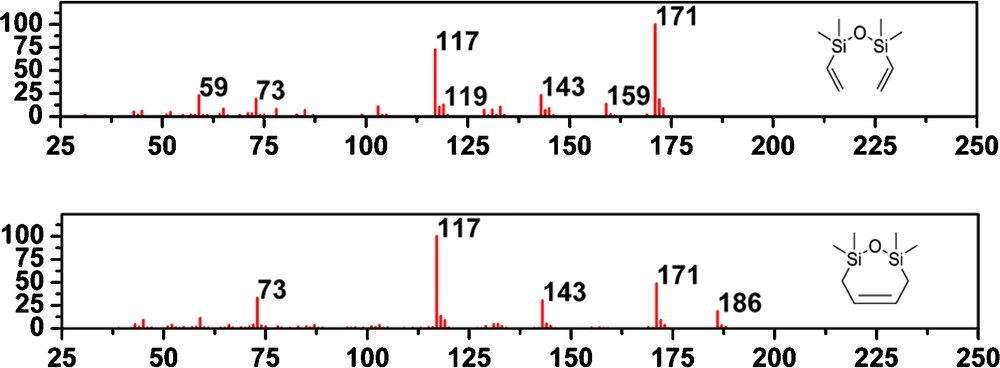
Comparison of mass spectra of the vinylsilane derivative 24 and the obtained compound 25.
The most significant difference between the spectrum of the compounds is the absence of the ion m/z = 159 for the silane obtained in this work. For the divinyldisiloxane 24, this ion is most likely produced by the silicon–carbon bond cleavage and the elimination of the ethene radical from the ion m/z = 186.
However, on account of the cyclic nature of the compound 25 showed on Fig. 8, such a bond cleavage will not lead to the formation of the ion m/z = 159. Cyclic structure of the silane present in the reaction mixture is supported by the higher intensity of the molecular ion m/z = 186 compared with the mass spectrum of the divinyldisiloxane 24. As it is showed on Fig. 9, the remaining peaks in the mass spectrum can be formed from both silanes. Therefore, the cyclic silane 25 showed on Fig. 8 is most likely present in the reaction mixture. Due to the short retention time of the investigated compound, it was reasonable to assume that the silane had relatively small molecular weight, and thus, we did not consider aggregates. The formation of both 24 and 25 in the presence of ruthenium catalysts can be explained. A vinylsilane derivative 24 can be formed if the double bond isomerisation and subsequent metathesis reaction occurred. Both reactions could be catalyzed by ruthenium species. Compound 25 is expected to be formed in the metathesis reaction of 12–15 in two subsequent metathesis processes. The first is the formation of ruthenium complex with silane chain and styrene derivative abstraction. The second includes intramolecular cyclization, which leads to the oxadisilepine derivatives and benzylidene ruthenium catalyst. Noteworthy the presence of 25 supports the intramolecular process operating in the investigated system.

Possible structures for the silane by-product.
3 Conclusions
A series of compounds including two styrenes connected by a siloxane linker (12–15) were synthesized and investigated in metathesis reactions catalyzed by selected ruthenium catalysts 1–3. Indenylidene ruthenium catalyst 2 (Umicore™ M2) was the most efficient in the metathesis of 12 and 15 and as a result (E)-stilbene derivatives were formed. In the case of 13 and 14, the presence of the ortho-alkoxide substituents in the aromatic ring led to the formation of Hoveyda–Grubbs type catalysts, which were essentially inactive in the studied process. Therefore no metathesis product could be obtained in this case. Finally, despite the presence of the linker both inter- or intramolecular metathesis processes operate for the studied substrates.
4 Experimental
4.1 General information
All reactions were performed under argon using standard Schlenk techniques. Solvents were distilled from standard drying agents and kept under argon. NMR spectra were recorded with a Varian 500. 1H and 13C NMR chemical shifts are listed in ppm using CDCl3 as a standard. Column chromatography: Merck silica gel (230–400 mesh).
Gas chromatography (GC) was performed using GC–MS-QP2010 Ultra gas chromatograph coupled to QP-5000 (Shimadzu) quadrupole mass spectrometer (MS); GC–MS was equipped with an AOC-5000 auto-sampler (Shimadzu). Separation of the studied compounds was carried out with HP-5ms (Agilent) chemically bonded fused silica capillary column (30 m × 0.25 mm, 0.25 μm). Carrier gas (He) was delivered at a flow rate of 36.8 mL/min, split ratio was 50:1 and the pressure was maintained constantly. Chromatographic conditions were as follows: injector temperature 300 °C; temperatures of the MS transfer line and ion source were set to 300 °C and 320 °C, respectively. The following temperature program of the column oven was used: initially 40 °C maintained for 2 min, then 12 °C/min to 300 °C, maintained for 10 min; analysis was completed in 33.7 min. MS was equipped with electron impact (EI) ion source (70 eV) and the spectrum was acquired in the total ion current (TIC) mode in the mass range of 35–500 m/z, solvent cut was 5 min. GC–MS solution 2.53 (Shimadzu) software was used to process the data.
To check the linearity of the MS detector response, a linear regression analysis of the peaks areas versus concentration of the studied compounds was carried out. The linearity was determined by the square correlation coefficients of the calibration curves that were generated by injections of the standard solutions at concentration levels between 0.1 and 0.5 mg/mL. The straight lines with the regression coefficients ≥ 0.99 were obtained.
4.1.1 Starting materials
Catalyst [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro-(3-phenyl-1H-inden-1-ylidene)(tricyclohexylphosphine)rutchenium 2 was purchased from UMICORE and used without purification. Catalyst bis(tricycylohexylphosphine)benzylidene rutchenium dichloride 3 was purchased from Aldrich and used without purification. Catalyst 1 was synthesized according literature [24].
4.2 Typical procedure for synthesis of 12–15 derivatives
A Schlenk tube was charged with an allylbenzene (1 mmol), TMEDA (1.2 mmol) and THF (10 mL) under an argon atmosphere. The solution was cooled to 0 °C. n-Buthyllithium (1.2 mmol; 2.5 mol/L solution in n-Hexane) was slowly added. Next, a cooling bath was removed and the reaction mixture was stirred at 25 °C for 1 h. Then, after cooling the reaction mixture to −40 °C, 1,3-dichloro-1,1,3,3-tetramethyldisiloxane (0.6 mmol) was added. Reaction proceeded at −40 °C for 30 min and then the reaction mixture was allowed to reach room temperature. The mixture was stirred at room temperature for 15 h, while the reaction progress was checked by TLC analysis. After that time, the solvent was removed from the reaction mixture by evaporation. Oily residue was purified by column chromatography (n-Hexane/Et2O 97:3). The (E)/(Z) ratio was determined by 1H NMR.
4.2.1 Compound 12: 1,3-dicinnamylo-1,1,3,3-tetramethyldisiloxane
Colorless oil, mixture of (E)- and (Z)-isomers, 92% yield, 1H NMR (500 MHz, CDCl3, 25 °C) δ = 0.12 (s, 6H), 1.71 (bd, 2H, J = 6.79 Hz); 6.16–6.28 (m, 2H); 7.12–7.18 (m, 2H), 7.22–7.32 (m, 3H) ppm; 13C NMR (125 MHz, CDCl3, 25 °C) δ = 0.2; 25.6; 125.5; 126.3; 126.7; 128.4; 129.0; 138.4 ppm; HRMS (ESI) calcd for [M+Na+] (C22H30OSi2Na): 389.1750; found: 389.1733; IR (film): ν 3024, 2956, 1253 (stretching Si–O–Si), 1064, 961, 851, 831, 799, 748, 692 cm−1
4.2.2 Compound 13: 1,3-di[3-(2-methoxyphenyl)allyl]-1,1,3,3-tetramethyldisiloxane
Colorless oil, mixture of (E) and (Z) isomer 91:9; 86% yield, 1H NMR (500 MHz, CDCl3, 25 °C) δ=0.21 (s, 6H); 1.83 (dd, 2H, J = 1.29 Hz, J = 8.25 Hz); 3.87 (s, 3H); 6.30 (dt, 1H, J = 15.84, J = 8.25 Hz); 6.66 (d, 1H, J = 15.84 Hz); 6.89 (dd, 1H, J = 0.8, J = 8.25 Hz); 6.95 (ddd, 1H, J = 0.65, J = 7.43, J = 7.6 Hz); 7.21 (ddd, 1H, J = 1.78, J = 7.43, J = 8.25 Hz); 7.43 (dd, 1H, J = 1.78, J = 7.6 Hz) ppm; 13C NMR (125 MHz, CDCl3, 25 °C) δ = 0.2; 26.1; 55.4; 110.8; 120.6; 123.6; 126.0; 127.2; 127.4; 127.6; 156.0 ppm; HRMS (ESI) calcd for [M+Na+] (C24H34O3Si2Na): 449.1944; found: 449.1935; IR (film): ν 2956, 1488, 1242 (stretching Si–O–Si), 1052, 1031, 806, 748 cm−1
4.2.3 Compound 14: 1,3-di[3-(2-isopropoxyphenyl)allyl]-1,1,3,3-tetramethyldisiloxane
Colorless oil, mixture of (E) and (Z) isomers 92: 8; 92% yield, 1H NMR (500 MHz, CDCl3, 25 °C) δ = 0.13 (s, 6H); 1.34 (d, 6H J = 6.15 Hz); 1.75 (dd, 2H J = 1.29, J = 8.24 Hz); 4.51 (Sept, 1H, J = 6.15); 6.22 (dt, 1H, J = 15.85, J = 8.24 Hz); 6.57 (d, 1H, J = 15.85 Hz); 6.83–6.91 (m, 2H); 7.11 (m, 1H); 7.37 (dd, 1H, J = 7.60, J = 1.61 Hz) ppm; 13C NMR (125 MHz, CDCl3, 25 °C) δ = 0.2; 22.3; 26.1; 70.7; 114.4; 120.7; 124.1; 126.2; 126.9; 127.0; 128.9; 154.4 ppm; HRMS (ESI) calcd for [M+Na+] (C28H42O3Si2Na): 505.2570; found: 505.2575; IR (film): ν 2976, 1484, 1238 (stretching Si–O–Si), 1119, 1062, 844, 803, 747 cm−1
4.2.4 Compound 15: 1,3-di[3-(3,4-dimethoxyphenyl)allyl]-1,1,3,3-tetramethyldisiloxane
Pale yellow oil, mixture of (E) and (Z) isomers 88:12; 90% yield. 1H NMR (500 MHz, CDCl3, 25 °C) δ = 0.13 (s, 6H); 1.69 (dd, 2H J = 0.97, J = 8.08); 3.86 (s, 3H); 3.87 (s, 3H); 6.08 (dt, 1H, J = 7.92, J = 8.09, J = 15.68), 6.19 (d, 1H, J = 15.67); 6.78 (d, 1H, J = 8.25); 6.82 (dd, 1H, J = 1.78, J = 8.91); 6.85 (d, 1H, J = 1.78) ppm; 13C NMR (125 MHz, CDCl3, 25 °C) δ = 0.2; 25.4; 55.8; 55.9; 108.4; 111.3; 118.2; 124.7; 128.6; 131.7; 147.9; 149.0 ppm; HRMS (ESI) calcd for [M + Na+] (C26H38O5Si2Na): 509.2155; found: 509.2138; IR (film): ν 2955, 1514, 1256 (stretching Si–O–Si), 1234, 1158, 1139, 1059, 1028, 836, 802 cm−1
4.3 Typical procedure for metathesis reaction
A Schlenk tube was charged witch disiloxane derivatives (0.25 mmol) and the catalyst (0.0125 mmol) in dry toluene (2,5 mL; or DCM) under an argon atmosphere. The mixture was stirred in an oil bath at 80 °C (or 40 °C, respectively). Reaction progress was checked by GC–MS analysis. Next, the solvent was removed on a rotary evaporator. Oily residue was purified by column chromatography (n-Hexane/Et2O 97:3), and the product was obtained as white crystals. The crude product was crystallized from toluene at −20 °C.
Metathesis of compound 12 yields product 16 as white crystals (67% yield). 1H, 13C NMR spectra were according to the literature [25].
Metathesis of compound 13 gives the product as white crystals of compound 19. 60% yield. 1H, 13C spectra were according to the literature [26].
4.4 Typical procedure for a stability test
A Schlenk tube was charged with disiloxane derivatives 12–15 (0.25 mmol) and dry toluene (2.5 mL) was added under an argon atmosphere. Next, a 10 μL sample of this solution was taken and diluted to 1 mL with the same solvent and analysed by GC–MS. The mixture was then stirred in an oil bath at 80 °C for 3 h. Next, the second GC–MS analysis was made in the same manner. No change was observed.
4.5 Catalyst 20 formation
Disiloxane derivative 12 (0.156 mmol), catalyst 2 (0.13 mmol) and copper(I) chloride (0.13 mmol) were added to the Schlenck flask in dry toluene (1.5 mL) under argon atmosphere. The mixture was stirred in an oil bath at 80 °C; progress of the reaction was controlled by TLC. Full conversion was observed after 15 min. The reaction mixture was cooled to room temperature, the solvent was removed in vacuo, and the product was purified by column chromatography (c-Hexane/Ethyl Acetate 4:1). Removal of the solvent, washing with a small amount of n-pentane and drying under vacuum afforded the product as a green solid in 49% yield. 1H and 13C NMR spectra were according to the literature [24].
Acknowledgements
Financial support was provided by the University of Warsaw (Grant No. 501/86-DSM-100100 and No. 501/86-DSM-102400). We wish to thank Dr. Hanna Majewska-Elżanowska for proof reading the manuscript.