

1 Introduction
Les britholites, correspondant à des apatites phosphosilicatées, font l’objet de nombreuses études à caractère aussi bien fondamental qu’expérimental, car depuis la découverte du site d’Oklo [1–3], elles sont considérées comme particulièrement prometteuses pour une utilisation en tant que matrice pour le confinement des déchets nucléaires. En effet, cette découverte a montré que ces matériaux peuvent retenir des radionucléides et certains produits de fission sur plusieurs millions d’années sans subir de dommages majeurs [4].
Plusieurs procédés ont été développés pour la préparation de ces matériaux contenant divers éléments, tels que les actinides, les lanthanides ou le césium [5–15]. La synthèse de britholites dopées avec ce dernier élément est usuellement réalisée par réaction à l’état solide à des températures de l’ordre de 1100 °C. De plus, la préparation au préalable de phases intermédiaires à des températures de l’ordre de 1400 °C est nécessaire. Par conséquent, il semble intéressant d’envisager la préparation de ces matériaux par mécanosynthèse, pour diminuer leur température de synthèse, d’autant que le césium est volatil aux températures élevées. Des travaux récemment rapportés dans la littérature [16–18] ont montré que cette méthode est efficiente pour la synthèse de ce type de matériaux.
La mécanosynthèse, initialement développée par Benjamin pour la préparation de composés métalliques [19], puis étendue par Yermakov et al. et Koch et al. à l’élaboration de divers types de matériaux [20,21], consiste à soumettre, dans un broyeur, un mélange de poudres à d’intenses chocs successifs, avec des fréquences réglables. Ces chocs induisent dans la matière des défauts ponctuels, linéaires ou de surface qui provoquent une augmentation de l’énergie libre du système. Pour réduire son énergie libre, différentes transformations s’y produisent et conduisent aux produits désirés [22].
Ainsi, cette méthode, mettant en œuvre une réaction à l’état solide à température ambiante, semble être attrayante. En effet, outre sa simplicité, rapidité et faible consommation d’énergie, elle se prête à une production industrielle. En outre, elle permet d’obtenir des poudres de grande surface spécifique, favorisant ainsi le frittage des matériaux.
Dans ce travail, nous envisageons la synthèse par mécanosynthèse d’apatites phosphosilicatées strontiques dopées avec du lanthane et du césium.
2 Protocole expérimental
2.1 Préparation des poudres
Les échantillons ont été synthétisés à partir de SrCO3 (> 96 % Riedel de Haen) ou SrO, La2O3 (> 99,5 % Prolabo), SiO2 (> 99,5 % Alfa), SrF2 (> 99,5 % Prolabo), Cs2CO3 (> 99 % Fluka), NH4F (> 95 % Merck) et Sr2P2O7.
2.1.1 Préparation de Sr2P2O7
Le pyrophosphate de strontium a été obtenu selon la réaction suivante :
| 2 SrCO3 + 2 (NH4)2HPO4 → Sr2P2O7 + 4 NH3 + 2 CO2 + 3 H2O | (1) |
Après avoir été broyé et homogénéisé dans un mortier en agate, le mélange de carbonate de strontium et d’hydrogénophosphate de di-ammonium a été mis en forme par pressage uniaxial à froid, puis calciné à 900 °C pendant dix heures. La montée en température a été effectuée à une vitesse de 10 °C/min.
2.1.2 Préparation des apatites au césium
Les apatites, de formule générale Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 avec x = 0, 1 et 6, ont été préparées selon deux protocoles différents.
Selon le premier mode opératoire, le broyage des réactifs a été effectué en une seule étape en utilisant, soit du carbonate de strontium :
| SrCO3 + (1+x)/2 La2O3 +1/2 Cs2CO3 + (6−x)/2 Sr2P2O7 + x SiO2 + SrF2→Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 + 3/2 CO2 | (2) |
soit de l’oxyde de strontium :
| SrO + (1 + x)/2 La2O3 + 1/2 Cs2CO3 + (6−x)/2 Sr2P2O7 + x SiO2 + SrF2 → Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 + 1/2 CO2 | (3) |
Dans le second mode opératoire, des phases apatitiques intermédiaires, de formule générale Sr8−xLa1+x(PO4)6−x(SiO4)xF, avec x = 0, 1 et 6, ont été préparées, dans une première étape, selon l’équation suivante :
| 3/2 SrO + (1 + x)/2 La2O3 + (6−x)/2 Sr2P2O7 + x SiO2 + ½ SrF2 → Sr8−xLa1+x(PO4)6−x(SiO4)xF | (4) |
Puis, dans une seconde étape, du carbonate de césium et du fluorure d’ammonium ont été ajoutés aux phases précédemment obtenues selon l’équation suivante :
| Sr8−xLa1+x(PO4)6−x(SiO4)xF +1/2 Cs2CO3 + NH4F → Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 + 1/2 CO2 + NH3 +1/2 H2O | (5) |
Avant la pesée des réactifs, l’oxyde de lanthane a été calciné à 1000 °C pendant 12 heures dans un four à moufle et le carbonate de césium a été chauffé dans une étuve à 80 °C pendant une nuit. L’oxyde de strontium utilisé (Eq. (3)) a été obtenu par calcination du carbonate de strontium à 1100 °C pendant 12 heures.
Les échantillons ont été préparés en utilisant un broyeur planétaire, de type Fritsch Pulvérisette 7. Les réactifs, pris dans les proportions stœchiométriques, ont été introduits dans une jarre de 45 cm3 contenant cinq billes de 12 mm de diamètre. Le rapport masse de la poudre sur masse des billes est 1:34. La vitesse de rotation du plateau et celle de la jarre sont, respectivement, égales à 500 et 1000 tr/min. Ces conditions opératoires correspondent à une énergie cinétique de choc de 0,151 J/choc, une fréquence de choc de 100 Hz et une puissance injectée de choc de 15,1 W/g. Les durées de broyage ont varié entre 30 minutes et 25 heures. Certains échantillons ont été calcinés sous un flux d’argon à 900 °C pendant 12 heures.
Dans les sections suivantes, les échantillons seront, respectivement, nommés Sr8LaCs, Sr7La2Cs et Sr2La7Cs pour x = 0, 1 et 6.
2.2 Caractérisation des poudres
L’analyse par diffraction des rayons X (DRX) des échantillons a été effectuée au moyen d’un diffractomètre des RX sur poudre PANalytical X’Pert Pro, en utilisant la radiation Kα du cuivre (Kα1 = 1,5406 Å). Les diffractogrammes ont été acquis dans l’intervalle 20–65° en 2θ avec un pas de 0,03° et un temps d’acquisition d’une seconde par pas. Le logiciel X’Pert HighScore Plus de la firme PANalytical a été utilisé pour identifier les phases cristallines par comparaison à la base de données ICDD PDF-2.
Les spectres d’absorption infrarouge ont été enregistrés à l’aide de deux spectromètres – EQUINOX 55 équipé d’un logiciel OPUS/IR et Perkin Elmer (SPECTRUM 100) – en utilisant la technique des pastilles de KBr.
3 Résultats et discussion
3.1 Poudres préparées
3.1.1 Diffraction des rayons X
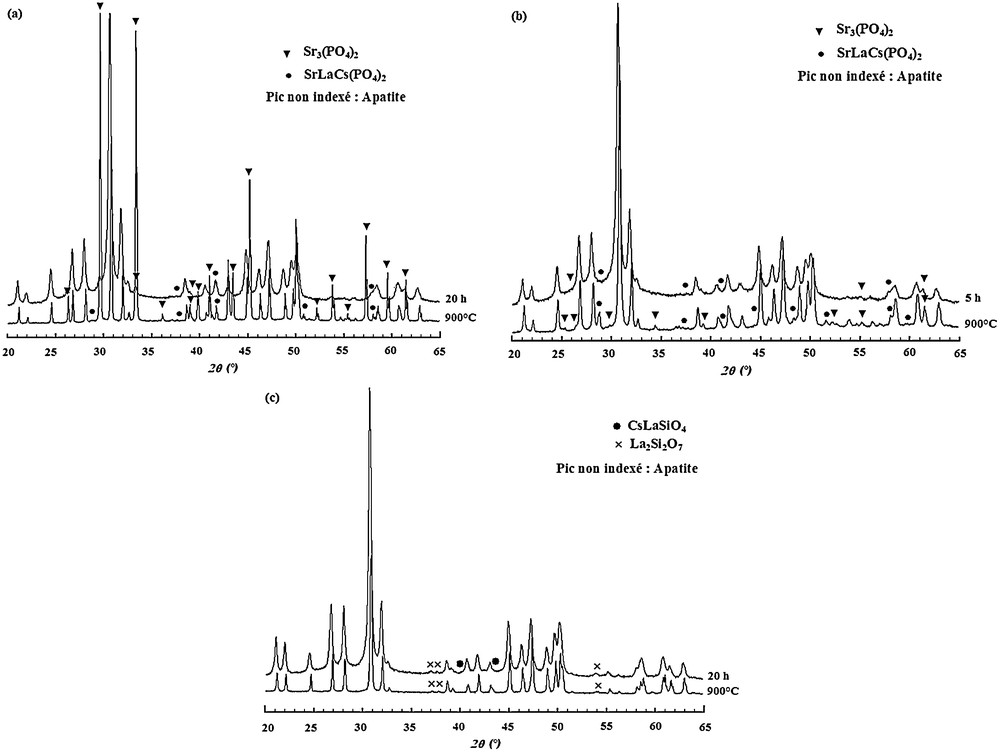
Les diagrammes DRX des trois échantillons préparés en une seule étape pour des durées de broyage comprises entre 30 minutes et 20 heures, selon l’Eq. (2), sont présentés sur la Fig. 1. Pour des durées de broyage de 30 minutes et une heure, les réactifs restent les phases majeures dans les échantillons. Ces phases, correspondant à SrCO3, La2O3, Cs2CO3, SiO2, Sr2P2O7 et SrF2, ont été indexées, respectivement, avec les fiches JCPDS no 01-074-1491, 01-074-2430, 00-035-0962, 01-085-0798, 00-024-1011 et 01-086-2418. À partir de trois heures de broyage, les diagrammes DRX révèlent, pour les trois échantillons, la formation d’une phase apatitique majoritaire, indexée avec la fiche JCPDS no 00-050-1744.

Diagrammes de diffraction des rayons X des échantillons préparés selon l’Eq. (2) : (a) : Sr8LaCs ; (b) : Sr7La2Cs ; (c) : Sr2La7Cs.
Le diffractogramme de l’échantillon Sr8LaCs, broyé pendant 30 minutes (Fig. 1a) montre l’émergence de quatre nouvelles raies DRX à 21,99 ; 31,83°; 38,54° et 42,87°, appartenant à la phase apatitique. Notons que l’intensité des raies DRX augmente avec la durée de broyage, alors que l’intensité des raies DRX des réactifs diminue. Il est à signaler que la majorité des réactifs disparaissent après une heure de broyage et que des pics de diffraction supplémentaires apparaissent. Ils ont été attribués aux composés Sr3(PO4)2 et SrLaCs(PO4)2, indexées, respectivement, à partir des fiches JCPDS no 01-085-0905 et no 00-035-0426. Ces phases restent présentes même pour des durées de broyage de 25 heures (Fig. 2a).

Diagrammes de diffraction des rayons X des échantillons préparés selon l’Eq. (3) : (a) : Sr8LaCs ; (b) : Sr7La2Cs ; (c) : Sr2La7Cs.
Pour l’échantillon Sr7La2Cs (Fig. 1b), préparé en une seule étape, les diffractogrammes des mélanges broyés pendant 30 minutes et une heure sont pratiquement identiques à ceux de l’échantillon Sr8LaCs. Pour des durées de broyage comprises entre trois et cinq heures, toutes les raies DRX appartiennent à la phase apatitique. Au-delà, il y a apparition de nouvelles raies associées aux phases Sr3(PO4)2 et SrLaCs(PO4)2. L’absence de détection de ces phases pour les durées de broyage comprises entre trois et cinq heures est probablement due à leurs faibles quantités ou à une mauvaise cristallisation.
Les diffractogrammes de la Fig. 1c correspondant à l’échantillon Sr2La7Cs montrent que pour 30 minutes et une heure de broyage, toutes les raies appartiennent aux produits de départ et que la phase apatitique n’apparaît qu’à partir de trois heures. Pour des durées de broyage égales ou supérieures à 10 heures, les diffractogrammes présentent, en plus de celles de l’apatite, de nouvelles raies correspondant aux phases secondaires La2Si2O7 et CsLaSiO4, indexées à partir des fiches JCPDS no 00-048-0052 et no 00-049-0663. Il est à noter que la silice reste présente jusqu’à une durée de broyage de 15 heures. Cette silice non incorporée a été remplacée dans la structure apatitique par les carbonates provenant des réactifs et qui ont été détectés par spectroscopie infrarouge (Section 3.1.2).
Afin de minimiser la quantité de carbonates incorporée dans la structure apatitique, nous avons réalisé des expérimentations en utilisant SrO au lieu de SrCO3, en mettant à profit le même protocole expérimental que précédemment. Les diagrammes DRX des trois échantillons broyés pendant 25 heures (Fig. 2) montrent qu’il y a formation des mêmes phases que celles observées en utilisant SrCO3.
De même, nous avons voulu vérifier l’influence du mode opératoire sur la nature des produits formés. Pour ce faire, nous avons utilisé le protocole expérimental préconisé dans la synthèse des apatites dopées au césium par réaction à l’état solide à haute température, c’est-à-dire en préparant, avant l’incorporation du césium, des phases apatitiques intermédiaires (Eq. (4)) [23]. La Fig. 3, présente les diagrammes DRX des apatites avant et après incorporation du césium, broyées, respectivement, pendant 25 heures. Pour les phases intermédiaires de formule Sr8−xLa1+x(PO4)6−x(SiO4)xF avec x = 0 et 1, les diffractogrammes révèlent, en plus de celle de l’apatite, la formation de Sr3(PO4)2 et Sr3La(PO4)3 (Fig. 3a et b). Pour x = 6, la phase formée en plus de l’apatite est La2Si2O7 (Fig. 3c). Après incorporation du césium, les phases détectées sont les mêmes que celles observées après broyage en une seule étape.

Diagrammes de diffraction des rayons X des échantillons préparés en deux étapes : (a) : Sr8LaCs ; (b) : Sr7La2Cs ; (c) Sr2La7Cs : (1) : apatites intermédiaires (Eq. (4)) ; (2) : apatites dopées au césium Eq. (5).
Le Tableau 1 présente les phases formées lors de la préparation des échantillons par mécanosynthèse (présent travail) et par réaction à haute température [24]. Ce tableau montre que la méthode de synthèse adoptée influence les phases formées après réaction. Boughzala et al. [24] ont observé après réaction, en plus de l’apatite, la phase SrLaCs(PO4)2, pour x = 0 et 1, et les phases CsLaSiO4 et Sr2SiO4, pour x = 6. En revanche, par mécanosynthèse, que la préparation soit réalisée en une seule ou en deux étapes, pour les deux premiers échantillons, le phosphate tri-strontique a été détecté en plus de l’apatite et de SrLaCs(PO4)2, alors que dans le cas du troisième échantillon, les phases secondaires sont CsLaSiO4 et La2Si2O7.
Produits formés par mécanosynthèse (présent travail) et par réaction à haute température [24].
| Mécanosynthèse | Réaction à haute température | ||
| Étape 1 | Étape 2 | ||
| Sr8LaCs | Apatite | Apatite | Apatite |
| Sr3(PO4)2 | Sr3La(PO4)3 | SrLaCs(PO4)2 | |
| SrLaCs(PO4)2 | Sr3(PO4)2 | ||
| Sr7La2Cs | Apatite | Apatite | Apatite |
| Sr3(PO4)2 | Sr3La(PO4)3 | SrLaCs(PO4)2 | |
| SrLaCs(PO4)2 | Sr3(PO4)2 | ||
| Sr2La7Cs | Apatite | Apatite | Apatite |
| La2Si2O7 | La2Si2O7 | Sr2SiO4 | |
| CsLaSiO4 | La2SiO5 | CsLaSiO4 |
Ainsi, d’après l’analyse par DRX, les seules phases renfermant du césium qui ont été formellement détectées sont SrLaCs(PO4)2 (x = 0 et 1) et CsLaSiO4 (x = 6). À ce stade, l’incorporation de cet élément dans la structure apatitique ne peut être affirmée avec certitude. Aussi, si cette insertion a effectivement eu lieu, la quantité insérée est nécessairement plus faible que celle introduite dans le mélange initial.
Les paramètres de maille des échantillons préparés avec SrCO3 et SrO ont été calculés à l’aide de la méthode des moindres carrés (Tableau 2). L’interprétation de l’évolution des paramètres avec la substitution reste difficile en raison des tailles différentes des ions en substitution (Tableau 3) [25]. Toutefois, il possible de tirer certaines conclusions. Quel que soit le réactif utilisé, le paramètre a augmente, alors que c diminue. Mais, cette diminution est très faible. Aussi, comme il est attendu, les paramètres de maille des apatites préparées avec SrCO3 sont plus faibles que ceux des apatites obtenues avec SrO, puisque ces dernières renferment normalement moins de carbonate. Par ailleurs, l’incorporation du césium dans l’apatite doit s’accompagner d’une augmentation importante du volume de maille. Le Tableau 4 montre que les paramètres des apatites intermédiaires sont plus faibles que ceux des apatites contenant du césium. Cela donne à penser qu’une partie de cet élément a été incorporée dans la structure apatitique.
Paramètres de maille des apatites obtenues en utilisant SrCO3 et SrO après un broyage de 25 heures.
| Échantillons | a = b (Å) | c (Å) | V (Å3) | |
| Sr8LaCs | SrCO3 | 9,726(4) | 7,288(3) | 597,718(3) |
| SrO | 9,728(5) | 7,287(4) | 597,881(4) | |
| Sr7La2Cs | SrCO3 | 9,730(3) | 7,285(4) | 597,963(3) |
| SrO | 9,739(5) | 7,287(4) | 599,234(4) | |
| Sr2La7Cs | SrCO3 | 9,747(2) | 7,277(3) | 599,395(2) |
| SrO | 9,751(3) | 7,270(2) | 599,310(2) |
Rayons ioniques des ions en substitution.
| Ions | Sr2+ | La3+ | Cs+ | P5+ | Si4+ | C4+ |
| Coordinence | 9 | 9 | 9 | 4 | 4 | 4 |
| Rayons ioniques (Å) | 1,31 | 1,22 | 1,78 | 0,17 | 0,26 | 0,15 |
Paramètres de maille des apatites intermédiaires.
| Échantillons | a (Å) | c (Å) | V (Å3) |
| Sr8LaF | 9,713(3) | 7,281(4) | 595,548(3) |
| Sr7La2F | 9,726 (5) | 7,279(5) | 596,979(5) |
| Sr2La7F | 9,738(3) | 7,263(5) | 597,248(4) |
3.1.2 Spectroscopie d’absorption infrarouge
La Fig. 4 présente les spectres d’absorption infrarouge des composés obtenus selon l’Eq. (2), après broyage à différentes durées. Ces spectres montrent l’avancement de la réaction de formation de la phase apatitique. Cependant, en raison de la complexité du système due aux réactifs utilisés, l’attribution des bandes observées a été parfois difficile. Pour tous les échantillons, les bandes situées vers 1453–1406 et 863 cm−1, associées au groupement CO32− [16,26,27], indiquent qu’une certaine proportion de carbonates provenant des réactifs a été incorporée dans la structure apatitique. Alors que les bandes observées vers 3444 et 1617 cm−1 sont attribuables à l’eau moléculaire adsorbée à la surface du solide [28]. L’attribution des bandes d’absorption des groupements PO4 et SiO4 a été effectuée par comparaison avec les spectres de phases apatitiques de compositions similaires [8–10,12,29–32].
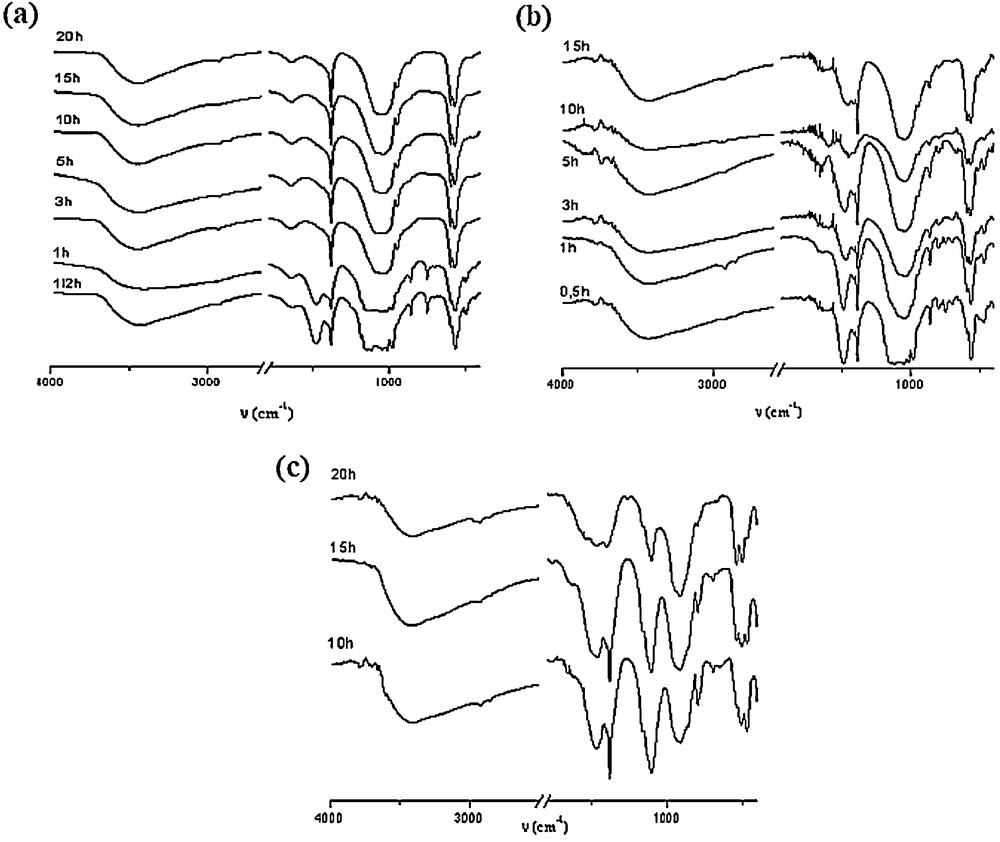
Spectres d’absorption infrarouge des échantillons broyés pendant des durées comprises entre 30 minutes et 20 heures (Eq. (2)) : (a) : Sr8LaCs ; (b) : Sr7La2Cs ; (c) : Sr2La7Cs.
Pour l’échantillon Sr8LaCs, après 30 minutes et une heure de broyage, les spectres montrent que la majorité des bandes d’absorption appartiennent aux réactifs (Fig. 4a). Ce n’est qu’à partir de trois heures que la formation de l’apatite devient évidente. Le spectre relatif à cette durée révèle les bandes caractéristiques des groupements PO43− dans un environnement apatitique. Ainsi, les bandes situées vers 1079 et 1032 cm−1 correspondent aux élongations asymétriques (ν3), la bande à 946 cm−1 est liée aux élongations symétriques (ν1) et les bandes vers 592 et 563 cm−1 sont relatives aux déformations asymétriques (ν4). Notons que les spectres restent pratiquement identiques jusqu’à 20 heures.
La Fig. 4b montre les spectres d’absorption infrarouge de l’échantillon Sr7La2Cs broyé entre 30 minutes et 15 heures. Comme pour Sr8LaCs, les bandes d’absorption observées, pour des durées de broyage de 30 minutes et une heure, sont relatives aux réactifs. Pour une durée de trois heures, le spectre révèle, en plus des bandes d’absorption correspondant aux groupements phosphate, des bandes associées aux groupements silicate, détectées vers 916 (ν3), 858 (ν1) et 468 (ν4) cm−1. En plus de ces bandes, on note la présence de bandes de faibles intensités vers 799, 745 et 699 cm−1 attribuées à la silice [33], non détectée par DRX, probablement en raison de sa faible quantité. Cette silice non incorporée a été remplacée dans la structure apatitique par les carbonates provenant des réactifs.
Les spectres de l’échantillon Sr2La7Cs, broyé pendant 10, 15 et 20 heures, présentés sur la Fig. 4c sont typiques d’une apatite silicatée. Les épaulements détectés vers 900 cm−1 sont attribuables au mode de vibration de valence symétrique (ν1) du groupement SiO4. Les bandes positionnées vers 920, 500 et 540 et 420, 440 et 460 cm−1 sont, respectivement, associées aux modes de vibration ν3, ν4 et ν2. De même, pour cet échantillon silicaté, on note la présence de bandes de faibles intensités vers 800, 722 et 698 cm−1 attribuable à la silice [33].
3.2 Calcination des poudres
Les échantillons préparés avec du carbonate de strontium ont été calcinés sous atmosphère d’argon à 900 °C pendant 12 heures après avoir été, respectivement, broyés pendant 20 heures (Sr8LaCs et Sr7La2Cs) et cinq heures (Sr2La7Cs). Pour ce dernier échantillon, cette durée a été retenue en raison de l’absence de phases secondaires dans la poudre broyée (Section 3.1.1). Les diagrammes DRX obtenus sont présentés sur la Fig. 5.
Diagrammes de diffraction des rayons X des échantillons préparés selon l’Eq. (2) et calcinés à 900 °C pendant 12 heures : (a) : Sr8LaCs ; (b) : Sr7La2Cs ; (c) : Sr2La7Cs.
Pour l’échantillon Sr8LaCs, la Fig. 5a donne à penser qu’au cours du traitement thermique, une partie de l’apatite s’est décomposée avec formation du phosphate tri-strontique. Dans la poudre broyée, sa quantité est très faible par rapport à celle de l’apatite, alors qu’après calcination, les raies DRX des deux produits sont, respectivement, d’égale intensité. Pour la phase SrLaCs(PO4)2, nous notons uniquement une amélioration de sa cristallinité. La décomposition de l’apatite devrait normalement s’accompagner par la formation d’autres phases, mais celles-ci n’ont pas été détectées. Dans le cas de Sr7La2Cs, pour des durées de broyage comprises entre trois et cinq heures, la seule phase détectée est l’apatite. Après traitement thermique, le diagramme DRX révèle la présence en très faible quantité des phases Sr3(PO4)2 et SrLaCs(PO4)2 (Fig. 5b). Quant à l’échantillon Sr2La7Cs, la Fig. 5c montre que l’influence de la calcination ne s’est traduite que par l’amélioration de la cristallinité de la poudre, les proportions des phases secondaires, La2Si2O7 et CsLaSiO4, n’ayant pratiquement pas varié.
L’obtention de l’oxyapatite La9,33(SiO4)6O2 par traitement thermique à haute température est le plus souvent accompagnée par la formation des phases secondaires La2SiO5 et/ou La2Si2O7 [34–36]. À titre d’exemple, cette dernière phase se forme à l’état amorphe dès 400 °C et cristallise progressivement au cours de la montée en température [37]. Une fois formée, il devient quasiment impossible de l’éliminer par calcination et obtenir l’oxyapatite pure [38]. Vidal et Bernal [39] ont montré que le mécanisme de sa formation est lié à des réactions sur la surface des grains de silice. Dès lors, il a été possible pour Béchade et al. [38] de préparer l’oxyapatite pure en utilisant de l’oxyde de lanthane très pur. En effet, c’est l’oxyde de lanthane hydraté qui réagit avec la silice pour donner La2Si2O7. Signalons que Rodriguez-Reyna et al. ont pu obtenir une oxyapatite pure par broyage à sec de La2O3 et SiO2 [17]. Dans le présent travail, nous avons observé la formation de Sr3(PO4)2, que la synthèse soit effectuée en une ou deux étapes, et il reste présent quelle que soit la durée de broyage. À haute température (1400 °C), Sr3(PO4)2 s’est aussi formé au cours de la préparation des apatites intermédiaires, mais il a complètement disparu après l’introduction du césium dans la deuxième étape (1100 °C) [24]. Il semble donc que les conditions opératoires peuvent être déterminantes dans la formation d’une phase donnée. Toutefois, des conditions expérimentales appropriées ne peuvent, à elles seules, expliquer la différence observée dans la nature des phases secondaires formées selon les deux méthodes (présent travail et par traitement thermique). En effet, des considérations thermodynamiques et/ou cinétiques sont sûrement derrière cette différence.
4 Conclusion
Des apatites phosphosilicatées dopées au lanthane et au césium de formule chimique Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 avec x = 0, 1 et 6 ont été préparées par mécanosynthèse selon deux protocoles expérimentaux différents. Selon le premier mode opératoire, tous les réactifs ont été introduits simultanément dans le broyeur, alors que, selon le second mode, des apatites intermédiaires ont été préparées avant l’introduction du césium. Après broyage, l’analyse des poudres obtenues par DRX et spectroscopie d’absorption infrarouge montre que le protocole expérimental n’a pas d’influence sur la nature des produits formés. Pour les échantillons Sr8LaCs et Sr7La2Cs, en plus de la phase apatitique, il y a formation des phases secondaires Sr3(PO4)2 et SrLaCs(PO4)2, alors que pour Sr2La7Cs, les phases secondaires sont La2Si2O7 et CsLaSiO4. La formation de ces phases donne à penser que la quantité de césium incorporée dans l’apatite est plus faible que celle introduite initialement dans les mélanges.