1 Introduction
Les peptoïdes sont des oligoamides artificiels constitués d’unités de glycines N-substituées introduits par Zuckermann et al. en 1992 [1]. La différence structurale par rapport aux peptides parents réside dans le fait que les chaînes latérales ne sont plus portées par les carbones Cα mais par les atomes d’azote des amides. Il en résulte un squelette achiral et la présence d’amides N,N-disubstitués qui confèrent aux peptoïdes des propriétés pharmacocinétiques intéressantes, en termes par exemple de stabilité métabolique [2] ou de perméabilité membranaire [3]. Mais l’avantage indéniable des peptoïdes sur de nombreuses autres familles de mimes peptidiques réside dans leur facilité de synthèse, notamment par la méthode dite « submonomère » [4], et la très grande diversité moléculaire, facilement accessible puisque celle-ci ne repose que sur l’utilisation d’un large panel d’amines primaires [5]. Le squelette peptoïde est donc tout à fait adapté à la préparation de banques combinatoires de ligands peptidomimétiques. De nombreuses bibliothèques de peptoïdes ont été générées puis testées afin d’identifier de nouvelles activités biologiques [6]. Par exemple, des tripeptoïdes ligands des récepteurs adrénergique ou opiacé avec des affinités nanomolaires ont pu être mis en évidence [7]. Suite à la découverte de leurs propriétés foldamériques par la mise en évidence de l’importance de la nature des chaînes latérales, des études ont montré le potentiel de ces nouveaux peptidomimétiques à mimer des interactions protéine-protéine notamment comme antibactériens, surfactants pulmonaires et anticancéreux [8]. De nouvelles applications sont maintenant envisagées dans le domaine des matériaux [1c,9]. La famille des peptoïdes n’est maintenant plus seulement limitée aux oligomères de glycines N-substituées. L’idée initiale, consistant à déplacer les chaînes latérales d’un peptide, des carbones Cα vers les atomes d’azote des amides, a été appliquée à d’autres oligoamides, non naturels cette fois-ci (Fig. 1). C’est le cas des β-peptides, déclinés en β-peptoïdes (oligomères de β-alanines N-substitués) [10]. D’autres familles de composés apparentés aux peptoïdes ont été rapportées récemment dans la littérature. Il s’agit des α,β-peptoïdes alternés [11], des arylopeptoïdes [12], des N-hydroxy [13] et N-alcoxy-peptoïdes [14] ou bien encore des azapeptoïdes [15].

Architectures peptoïdes inspirées des peptides. En rouge : éléments structuraux présents dans le monomère de glycine N-substituée qui caractérise les peptoïdes. En bleu : modifications structurales apportées au niveau du squelette et des chaînes latérales.
Le premier objectif de cette revue est de faire une mise au point bibliographique sur ces nouvelles familles de composés apparentés aux peptoïdes. Nous décrirons successivement leurs méthodes de synthèse et l’état des connaissances sur leurs préférences conformationnelles. Les α-peptoïdes sont capables d’adopter des conformations privilégiées, notamment hélicoïdales, cependant une hétérogénéité conformationnelle est dans la plupart des cas observée. L’équilibre cis/trans des liens amides N,N-disubstitués est la raison principale de cette hétérogénéité. Cette revue dresse également un bilan sur les avancées récentes concernant le contrôle de la conformation des amides de peptoïdes, notamment grâce à l’établissement d’interactions locales non covalentes entre le squelette et les chaînes latérales.
2 Peptoïdes et familles de composés apparentés
À l’origine, les premiers oligomères α-peptoïdes longs d’une dizaine de résidus ont été obtenus par synthèse peptidique sur support solide en utilisant comme monomères, des glycines N-substituées protégées par un groupement Fmoc (Fig. 2a) [1a,16]. Le squelette étant achiral, l’épimérisation liée au couplage peptidique n’est plus un problème, en revanche celui-ci est rendu plus difficile par l’encombrement de l’amine secondaire. Les réactifs de couplage PyBop® ou PyBrOP® ont permis d’obtenir des résultats satisfaisants, cependant cette méthode de synthèse nécessite la préparation préalable de quantités importantes de monomères, ce qui la rend peu adaptée pour l’élaboration de bibliothèques de composés.

Les principales méthodes d’accès aux α-peptoïdes sur support et en solution.
Une seconde méthode plus efficace de synthèse supportée adaptée aux peptoïdes, appelée submonomer solid-phase synthesis a rapidement supplanté la première (Fig. 2b) [4]. C’est une méthode itérative en deux étapes. Une première étape permet la formation du squelette par couplage de l’acide bromoacétique avec l’extrémité N-terminale de l’oligomère porté par la résine. Une seconde étape de substitution nucléophile du bromoacétamide intermédiaire par une amine primaire permet d’introduire la chaîne latérale. Ces deux étapes sont répétées en faisant varier les amines primaires dont l’accessibilité permet une grande diversité de structure des peptoïdes. Une revue récente très complète de Culf et Ouellette répertorie les différentes méthodes « monomère » et « submonomère », les résines et les amines employées [5]. Il est intéressant de noter que les deux étapes de la méthode submonomère supportée peuvent être activées par micro-ondes ce qui permet un gain important en temps de réaction et en pureté des produits finaux [17]. Ce mode d’activation permet d’augmenter l’efficacité du couplage d’amines stériquement encombrées ou électroniquement désactivées. La méthode submonomère est également adaptée à la synthèse en solution d’oligomères de longueur inférieure à dix résidus à l’échelle du gramme. En solution, la première étape est une acylation de l’amine par le bromure de bromoacétyle [11]. Par la méthode submonomère supportée, des hétéro-oligomères allant jusqu’à 48 résidus de longueur ont pu être synthétisés [18]. De longs oligomères peuvent également être obtenus par polymérisation de N-carboxyanhydride (NCA) initiée par une amine primaire [19] ou plus efficacement par l’utilisation d’un carbène hétérocyclique (NHC) comme initiateur (Fig. 3) [20]. De manière plus anecdotique, la résistance des peptoïdes face aux protéases a permis d’envisager leur synthèse par bio-ligation de fragments catalysée par la clostripaïne conduisant à des oligomères allant jusqu’au 150-mères [21].

Polymérisation zwittérionique induite par carbène hétérocyclique.
2.1 Changements portant sur le squelette
2.1.1 β-peptoïdes
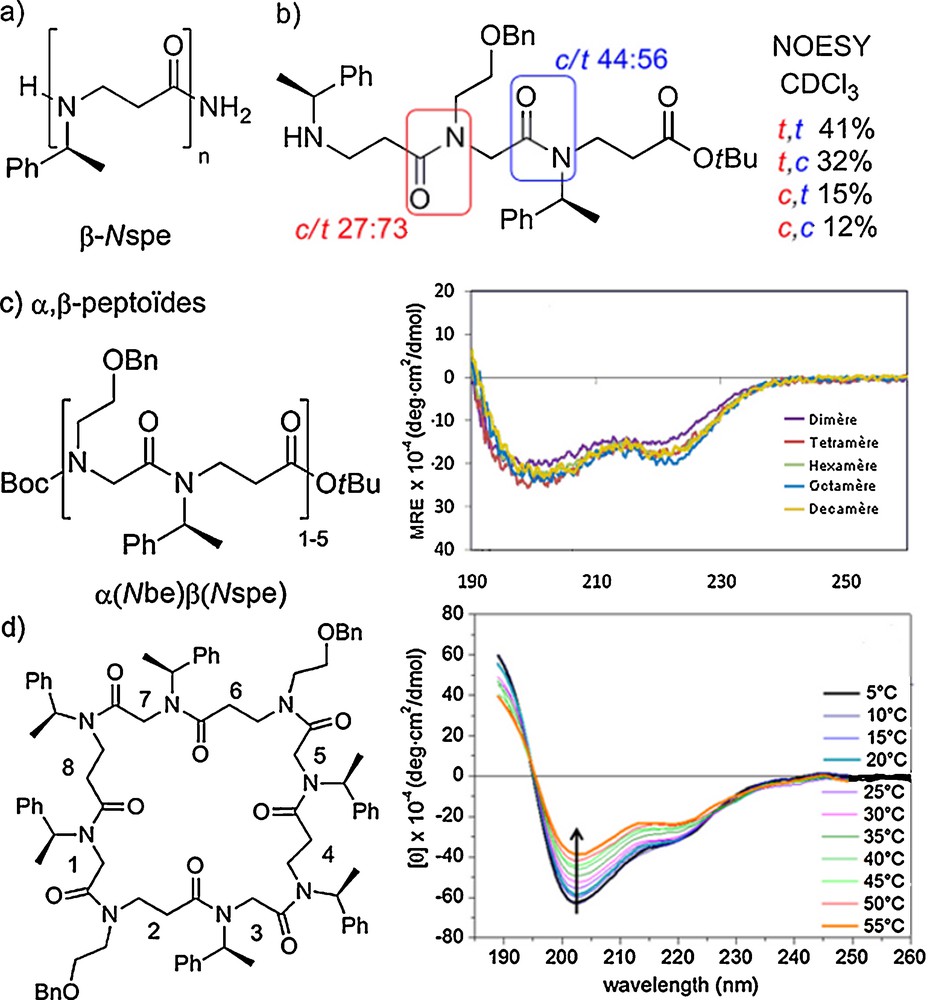
La méthode submonomère a été adaptée pour permettre l’accès aux β-peptoïdes, homologues des α-peptoïdes. Il s’agit également d’une méthode itérative en deux étapes : le squelette du β-peptoïde est formé par acylation de l’extrémité N-terminale avec le chlorure d’acryloyle puis la chaîne latérale est introduite par une réaction d’aza-Michael d’une amine primaire sur l’intermédiaire acrylamide (Fig. 4) [10a].
Synthèse des β-peptoïdes par la méthode submonomère (a) supportée, (b) en solution.
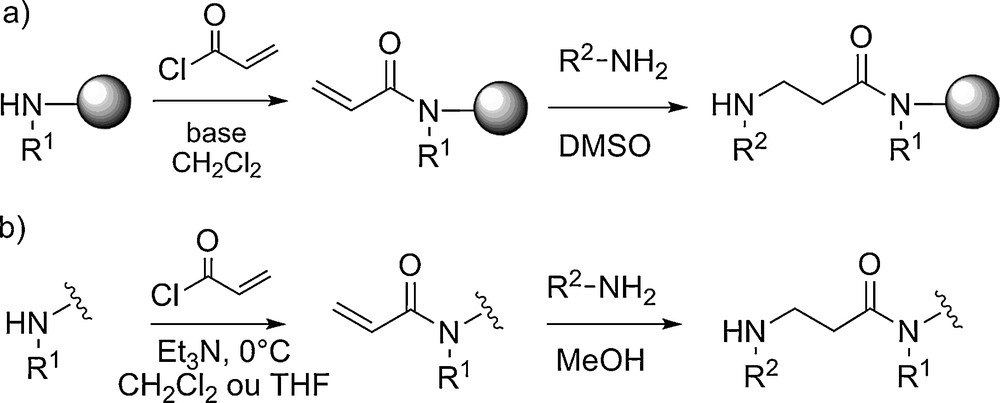
Les conditions de synthèse supportée des β-peptoïdes ont été beaucoup moins étudiées et optimisées que celles des α-peptoïdes. À l’heure actuelle elles ne permettent pas la synthèse de longs oligomères avec une pureté et un rendement suffisant. Pour remédier à ce problème, Shuey et al. ont utilisé une approche supportée par couplage de blocs (dimères ou trimères β-peptoïdes) [22]. Des oligomères longs de 18 unités ont ainsi pu être obtenus. Les auteurs ont également montré que la synthèse en solution par la méthode submonomère était le moyen le plus efficace pour obtenir les « blocs » en grande quantité. Par ailleurs, une optimisation des conditions réactionnelles en solution ainsi que l’utilisation d’amines primaires volatiles a permis la préparation de tétramères à l’échelle du gramme en sept étapes sans purification chromatographique intermédiaire (Fig. 5) [23]. Il faut noter que la modification des fonctions alcène, alcyne et amine tertiaire portées par les chaînes latérales, après assemblage du squelette (post-modification), permet l’introduction d’une grande diversité structurale.
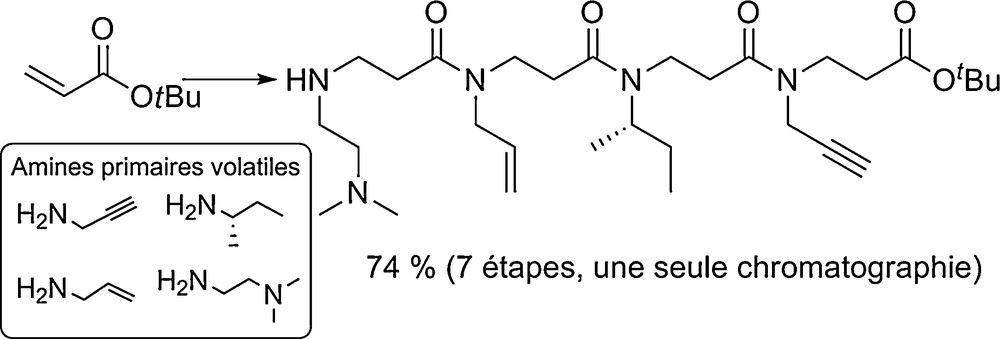
Synthèse des β-tétrapeptoïdes par la méthode submonomère utilisant des amines volatiles.
Récemment, la méthode monomère supportée a été exploitée pour la préparation d’un 16-mère en utilisant DIC/HOAt comme agent de couplage [24].
2.1.2 α,β-Peptoïdes
Les α,β-peptoïdes ont été introduits en 2009. Seule une méthode de synthèse en solution a été développée [11]. Elle combine les méthodes submonomères des α- et β-peptoïdes pour la construction d’oligomères α,β-alternés allant jusqu’à six résidus. Les plus longs oligomères sont obtenus par couplage peptidique de blocs. Comme en série β, l’accès sans chromatographie intermédiaire à des α,β-tétrapeptoïdes à partir d’amines volatiles a également été mis au point [23].
2.1.3 Arylopeptoïdes
L’insertion d’un groupement aryle dans le squelette des peptoïdes permet d’obtenir des aminométhylbenzamide N-alkylés. Ces oligomères ont été décrits pour la première fois par Zuckermann à Chiron Corporation dans une série de brevets1 puis par le groupe de Lokey [12a]. Uniquement des pentamères avaient été préparés par synthèse submonomère supportée utilisant des acides halogénométhylbenzoïques, ce qui n’avait pas permis d’étude approfondie de ce squelette, notamment d’un point de vue conformationnel. De récentes améliorations des voies d’accès à ces oligomères en solution et sur support permettent maintenant l’élaboration de bibliothèques d’arylopeptoïdes en série méta et para allant jusqu’aux 12-mères (Fig. 6) [12b,25]. Les premiers résultats concernant les propriétés de structuration de ces oligoamides aromatiques dépourvus de NH d’amides sont encourageants. Une première synthèse submonomère a été mise au point utilisant, une étape de couplage de l’acide chlorométhylbenzoïque à l’aide de COMU, un sel d’uronium dérivé de l’oxyma (2-cyano-2-(hydroxyimino) acétate d’éthyle) [25a]. Bien que très efficace, cette méthode s’est montrée peu adaptée pour l’introduction de chaînes latérales encombrées de type (S)-1-phényléthyle (spe) ou tBu. L’utilisation de chlorure d’acides dans l’étape d’acylation a permis de remédier à ce problème (Fig. 6) [25b]. Des résidus peptoïdes incorporant des hétérocycles aromatiques de type pyrazine, furane, oxazole et thiazole ont été récemment introduits dans des oligomères α-peptoïdes [26].
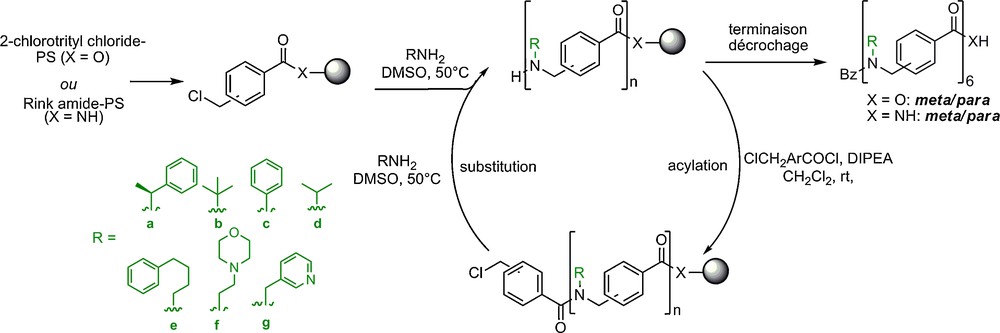
Synthèse supportée des arylopeptoïdes par la méthode submonomère.
2.2 Changements portant sur la substitution des azotes d’amides
2.2.1 N-hydroxy/alcoxy-peptoïdes et azapeptoïdes
Ces deux familles de peptoïdes ont été préparées selon la méthode submonomère en utilisant des hydroxylamines O-substituées lors de l’étape de substitution. Les temps de réaction des substitutions nucléophiles sont plus longs en raison d’une plus faible réactivité des hydroxylamines utilisées, par rapport à celle des amines servant habituellement à la synthèse des peptoïdes. La synthèse est décrite en solution pour les oligomères N-hydroxy [13] (Fig. 7a) et en phase solide pour les oligomères N-alcoxy [14] (Fig. 7b). Les N-hydroxy-peptoïdes sont obtenus dans un premier temps sous forme benzylée et une hydrogénolyse en présence de Pd/C sous pression atmosphérique permet d’obtenir les motifs N-hydroxy-peptoïdes. Dans le cas de la synthèse supportée des N-alcoxy-peptoïdes, des conditions douces de coupure de la résine Rink amide sont nécessaires (TFA à 7,5 %) afin d’éviter la coupure de la chaîne latérale.

N-hydroxy-, alcoxy-peptoïdes et azapeptoïdes. a : synthèse des N-hydroxy-peptoïdes ; b : synthèse des N-alcoxy-peptoïdes ; c : exemple de trimère azapeptoïde.
Les azapeptoïdes2 constitués d’unités acylhydrazides glycines ont été proposés récemmment par Kodadek et al. [15a] (Fig. 7c). Leur synthèse supportée est réalisée par la méthode submonomère en utilisant des acylhydrazides dans l’étape de substitution. Une bibliothèque de 10 000 tétramères a été préparée sur une résine Tentagel selon une approche split and pool. La structuration des azapeptoïdes n’a pas encore été étudiée. Il a seulement été montré que les amides adoptent très préférentiellement la géométrie trans, aussi bien dans le squelette qu’au niveau des chaînes laltérales. Les unités « peptoïdes hydrazides » ont déjà été envisagées comme substitut de proline dans un peptide, mais leur oligomérisation n’est pas encore rapportée [27].
3 Contrôle de l’isomérie cis/trans des peptoïdes
3.1 Hétérogénéité conformationnelle des oligo-peptoïdes
3.1.1 Énergétique de l’isomérie cis/trans des amides N,N-disubstitués
Les peptoïdes sont caractérisés par une flexibilité de squelette importante qui s’explique par :
- • l’absence de substituant sur les carbones Cα ;
- • la capacité réduite du squelette peptoïde à établir des liaisons hydrogènes intramoléculaires ;
- • la présence d’amides N,N-disubstitués qui sont en équilibre entre les formes cis et trans.
À cet égard on peut faire une analogie entre les peptoïdes et les séquences peptidiques contenant des prolines (Fig. 8a). Ces dernières forment un amide N,N-disubstitué avec le résidu qui les précède. La majorité des protéines contenues dans la PDB contiennent au moins un amide cis, et sur l’ensemble, 5,7 % des amides Xaa-Pro sont dans la conformation cis [28a]. Dans les deux cas, peptoïdes et amides Xaa-Pro, l’isomérie cis/trans s’explique en partie par une moins grande différentiation stérique entre les conformations cis et trans, en comparaison de celle qui existe dans le cas des amides des peptides. Les chaînes alkyles α-branchées conduisent par exemple à des proportions équivalentes d’amides cis et trans (Kcis/trans ∼ 1). La chaîne latérale phényléthyle (pe) très largement employée dans le domaine des peptoïdes induit une légère préférence pour la géométrie cis en raison d’effets stéréoélectroniques (vide infra) (Fig. 8b).
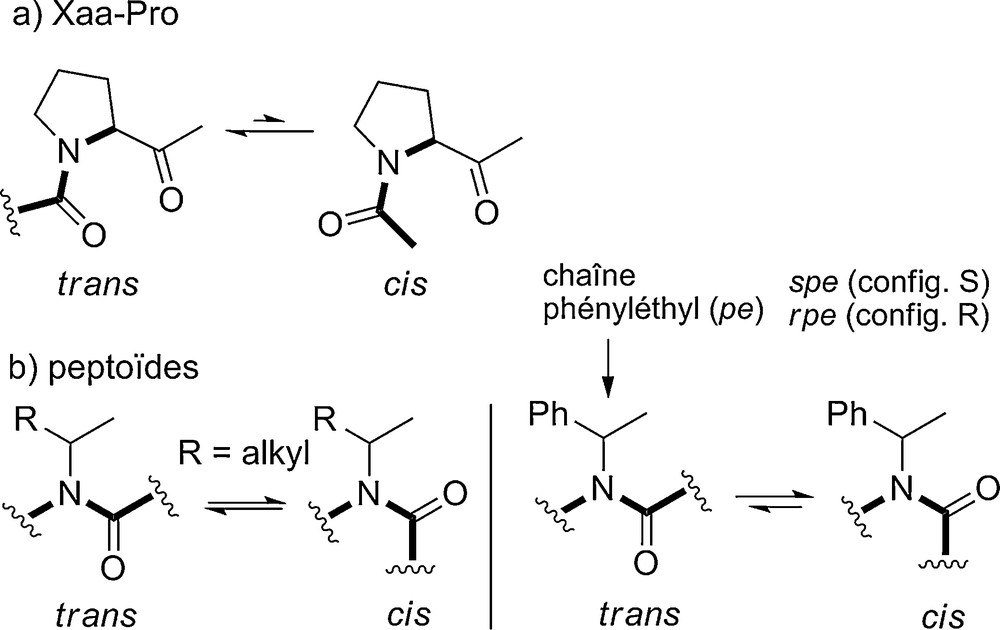
Isomérie cis/trans des amides N,N-disubstitués Xaa-Pro et des peptoïdes.
La cinétique d’isomérisation cis/trans étant lente à l’échelle de temps des expériences RMN, les spectres RMN de peptoïdes, en général très complexe, résultent de la combinaison des spectres des différents rotamères [28b]. L’isomérie cis/trans est certes responsable d’une hétérogénéité conformationnelle, mais le squelette polyglycine des peptoïdes possède néanmoins une propension à adopter des conformations préférentielles typiques qui sont présentées ci-après.
3.1.2 Structures secondaires adoptées par les α-peptoïdes
Les premières études portant sur la structuration des peptoïdes furent menées à partir de séquences portant exclusivement des chaînes latérales α-branchées chirales. L’hélice de type polyproline I (PPI) avec tous les liens amides cis, trois résidus par tour d’hélice et un pas d’environ 6 Å, est une conformation majoritairement rencontrée pour ce type de séquences ; l’hélicité droite ou gauche étant dictée par la stéréochimie de la chaîne latérale. Cette structuration a été initialement prédite [29] puis démontrée à partir d’homooligomères portant des chaînes latérales aromatiques. Par exemple, l’homo-pentamère (Nspe)5 constitué de résidus (S)-N-(1-phényléthyl)glycines forme majoritairement en solution une hélice droite [30]. Les oligopeptoïdes portant des chaînes latérales aliphatiques α-chirales adoptent également une structure hélicoïdale. Une analyse par diffraction des RX a clairement mis en évidence une conformation de type PPI, d’hélicité gauche pour un pentamère constitué de résidus (R)-N-(1-cyclohexyléthyl)glycines (Nrch) (Fig. 9a) [31]. Le spectre de dichroïsme circulaire (DC) de ce pentamère dans l’acétonitrile, bien que d’intensité relativement faible, est également caractéristique de l’hélice PPI avec un maximum vers 210 nm et deux minima vers 200 et 225 nm. La faible intensité du signal dichroïque révèle en fait une hétérogénéité conformationnelle en solution, provenant de l’isomérie cis/trans des liens amides (Kcis/trans ≈ 1 dans l’acétonitrile, mesuré par RMN 2-D). Il faut noter qu’une hétérogénéité du même ordre de grandeur caractérise le pentamère (Nspe)5 à chaînes latérales aromatiques. Une étude systématique par DC des homooligomères (Nrpe) comportant de trois à 20 résidus a permis de mettre en évidence un effet coopératif : le caractère hélicoïdal s’accroît jusqu’à 12 résidus [32]. La séquence du peptoïde est particulièrement importante dans le cas d’oligomères courts, comme l’ont montré Barron et al. sur une série d’hexamères [33a] : l’hélicité est favorisée lorsque les séquences sont constituées d’au moins 50 % de résidus à chaînes latérales chirales et aromatiques, ou bien lorsque l’hélice présente une ou plusieurs faces aromatiques, ce qui est le cas lorsque les chaînes aromatiques sont positionnées tous les trois résidus (position i, i+3). La présence d’une chaîne latérale α-chirale à l’extrémité C-terminale est également un facteur favorable. Le groupe de Barron a également montré dans le cas d’un oligopeptoïde hydrosoluble (36-mère) que la structure hélicoïdale était relativement insensible à l’environnement, que ce soit la nature du solvant (eau, MeOH, TFE), une élévation de température ou encore la présence d’un agent dénaturant tel que l’urée [33b].

Conformations de type PPI. a : représentation d’un pentamère (Nrch) et sa structure RX (hélicité gauche M) ; b : représentation d’un tétramère (N1npe) et sa structure RX (hélicité droite P).
La construction d’hélices de type PPI homogènes et stables en solution n’a été rapportée pour la première fois qu’en 2011 par Blackwell et al. [34]. Ce résultat unique repose sur l’utilisation de la chaîne latérale N-1-naphthyléthyle (1npe) très encombrée stériquement, qui a été incorporée dans des homo-oligomères de taille variable (1–13 mers) (Fig. 9b). L’homogénéité conformationnelle, appréciée grâce au rapport global Kcis/trans est déjà très élevée pour le dimère Ac-(s1npe)2-COOtBu (Kcis/trans = 16) et est totale dès le tétramère Ac-(s1npe)4-COOtBu (Kcis/trans > 19). La cristallographie des quatre premiers membres de la famille (monomère-tétramère) a permis de vérifier à l’état solide, la conformation cis des amides, et la concordance des angles dièdres avec ceux de structures PPI de peptides.
La conformation « threaded loop » caractérisée pour la première fois en 2006 [35], est spécifique de nonamères et repose sur l’établissement de quatre liaisons hydrogène intramoléculaires impliquant les carbonyles des résidus 2, 5, 7 et 9 et les extrémités N-terminale (sous forme d’ammonium) et C-terminale (CONH2) (Fig. 10a). Dans cette conformation, le peptoïde est replié sur lui-même et forme une boucle approximativement plane qui comporte autant de liaisons amide cis (c) que trans (t), selon la séquence c-c-t-t-t-c-t-c. Cette conformation atypique est particulièrement stable dans le cas de chaînes latérales chirales aromatiques phényléthyle (pe) [32] les composés à chaînes aliphatiques, tels que H-(sch)9-CONH2 et H-(ssb)9-CONH2 se structurent de façon similaire mais avec un caractère dynamique plus marqué (Fig. 10b). Dans les solvants protiques, tels que le méthanol et le trifluoroéthanol, la conformation en boucle disparaît au profit d’une structure hélicoïdale. Des études complémentaires ont montré que la conformation en boucle pouvait être stabilisée au dépend de l’hélice, et inversement, par l’incorporation en des positions clés de la séquence de chaînes latérales aromatiques électrodéficitaires [36].
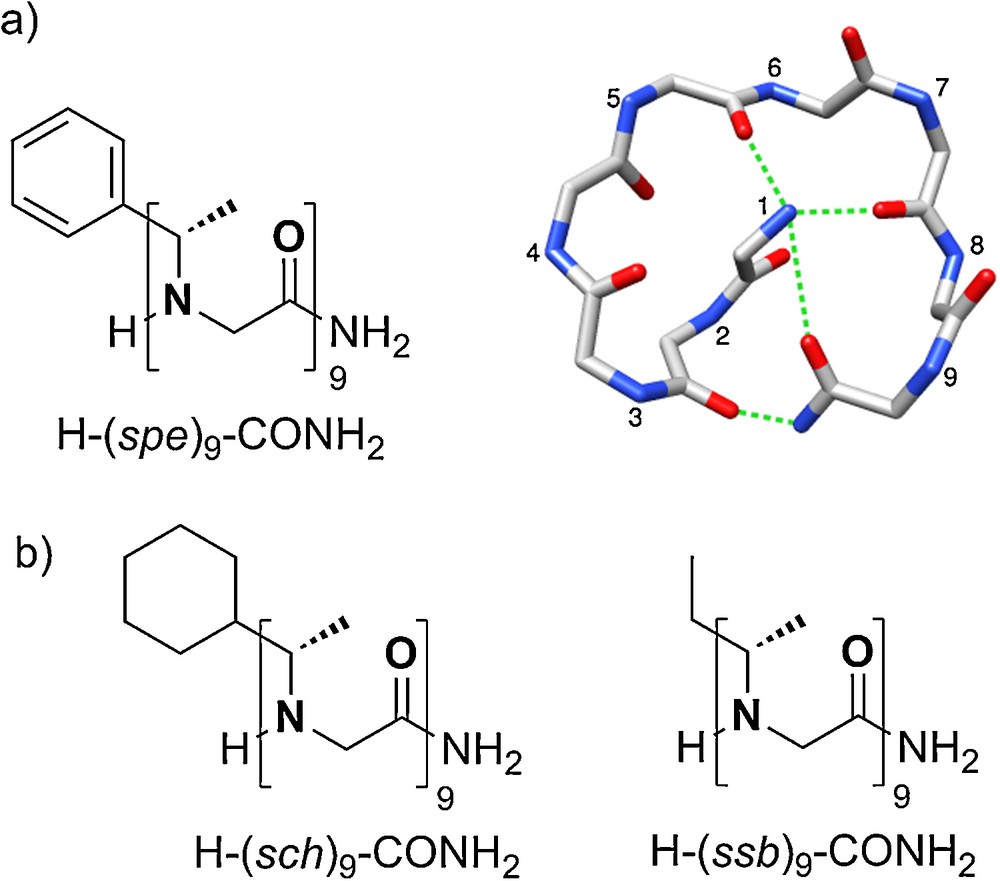
Conformation en boucle threaded loop. a : conformation établie par RMN à partir du nonamère H-(spe)9-CONH2. Les chaînes latérales sont omises pour plus de visibilité du modèle ; b : autres nonamères étudiés.
La conformation hélicoïdale de type PPI est la conformation préférentiellement adoptée par les peptoïdes. La structure de type polyproline II (PPII), dont tous les liens amides sont trans est néanmoins également rencontrée avec les peptoïdes. Les travaux de Kirshenbaum et al. ont montré que cette conformation était envisageable dans le cas d’oligomères constitués d’unités N-Arylglycines [37]. Les amides substitués par des groupes aryle montrent en effet une préférence marquée, souvent supérieure à 90 %, pour la conformation trans. Cette préférence, étudiée sur des modèles de type N-méthylacétanilide serait le résultat d’une répulsion électronique entre les groupes phényle et carbonyle au sein d’un même amide (Fig. 11a) [38]. Le contrôle de la conformation trans par les chaînes aryle a pu être vérifié au sein de structures cristallines de dimères et de macrocycles portant des chaînes alkyle et phényle. C’est une étude par modélisation moléculaire d’un hexamère N-(phényl)glycine qui a permis de montrer que la structure PPII, avec environ 3,1 résidus par tour et un pas de 9 Å, était énergétiquement favorable. En l’absence totale de carbone chiral, le sens de l’hélice n’est évidemment pas contrôlé. Un phénomène d’atropoisomérie est cependant observé pour des N-aryl peptoïdes portant des groupes phényle ortho-substitués (tBu, I), dont la rotation (angle dièdre χ1) est contrainte (Fig. 11b). Cette stratégie a permis au groupe de Kirshenbaum de réaliser une énantioséparation d’anilides modèles [39].

N-Aryl glycines. a : équilibre rotamérique déplacé vers la forme trans ; b : atropoisomérie des N-arylglycines ortho-substituées.
3.1.3 Préférences conformationnelles des β- et α,β-peptoïdes
Aucune structure secondaire n’a encore pu être mise en évidence pour les β- et α,β-peptoïdes. L’étude de leur structuration se limite essentiellement à des analyses par dichroïsme circulaire. Indépendamment l’un de l’autre, les groupes d’Arvidsson [40] et d’Olsen [41] ont étudié par DC des homooligomères β-peptoïdes présentant des chaînes spe (Fig. 12a), afin de pouvoir les comparer à des spectres de référence en série α-peptoïde. Les spectres DC à double minima (∼200 et 220 nm) montrent une similitude avec ceux des α-peptoïdes Nspe qui adoptent la conformation PPI. Cette similitude pourrait être le signe d’une structuration hélicoïdale comme prédit par Hofmann et al. [42a], mais en l’absence de données à haute résolution3, la seule certitude est que les β-peptoïdes linéaires adoptent en solution un ensemble de conformations privilégiées. Les α,β-peptoïdes ont également été surtout étudiés par DC en raison de la complexité des spectres RMN proton, engendrée par l’hétérogénéité conformationnelle cis/trans [11,43]. La proportion cis/trans des amides a pu être déduite d’expériences NOESY sur de courts oligomères (Fig. 12b). Les spectres DC des oligomères α,β-peptoïdes présentant de façon alternée 50 % de chaîne spe et 50 % de chaîne benzyloxyéthyle (be) (Fig. 12c), montrent également de fortes similitudes avec ceux des α-peptoïdes Nspe hélicoïdaux. Ils sont caractérisés par deux maxima négatifs à 200 et 222 nm et une intensité du signal dichroïque comparable. En revanche, en contraste avec les α-peptoïdes Nspe, ni effet coopératif, ni même aucun effet de température ne sont observés pour la famille α(Nbe)β(Nspe) linéaire. Ces résultats s’expliquent peut être en partie par la séquence α(Nbe)β(Nspe) qui ne présente qu’une chaîne latérale chirale sur deux. Des études complémentaires doivent maintenant être menées sur des séquences α(Nspe)β(Nspe) ne présentant que des chaînes chirales. L’influence du nombre de chaînes latérales chirales et de leur positionnement sur les unités α ou β a été étudiée sur des octamères en série cyclique [44]. L’un des composés (Fig. 12d) possédant six chaînes spe et deux chaînes be positionnées sur des unités β opposées dans le cycle a permis l’obtention du premier cliché de diffraction des RX de la famille α,β-peptoïde. Il est à noter que ce même composé possède un spectre RMN 1H mieux résolu que ceux de composés apparentés et que son signal dichroïque, plus intense que celui de son homologue linéaire est sensible à une élévation de température.
3.1.4 Structures secondaires adoptées par les N-hydroxy et N-alcoxy-peptoïdes
Les substitutions N-hydroxy [13] ou N-alcoxy [14] permettent un excellent contrôle de la géométrie des amides correspondants. Des études RMN sur le monomère N-hydroxy Ac-NOH-Pip (CDCl3) et les homodimères Ac-(NOH)2-Pip (CDCl3) et Ac-(NOrpe)2-NH2 (CD3CN) ont montré le caractère trans des amides pour ces peptoïdes (Fig. 13). Cette préférence s’expliquerait par des considérations stériques, l’oxygène des N-hydroxy/alcoxyamides crée moins d’encombrement qu’un groupe alkyle. Par ailleurs, une répulsion électronique entre l’oxygène lié à l’azote et le carbonyle de l’amide défavorise la conformation cis. Dans le cas du monomère Ac-NOH-Pip, le déplacement chimique du proton NO-H à 8,26 ppm dans CDCl3 montre son implication dans une liaison H qui pourrait s’établir soit de façon intramoléculaire comme montré sur la Fig. 13, soit de façon intermoléculaire, comme révélé par DRX à l’état solide. Le dimère Ac-(NOH)2-Pip forme des agrégats dans CDCl3, même à de faibles concentrations. À l’état solide un arrangement de type feuillet antiparallèle est observé (Fig. 14).

Exemples de N-hydroxy et N-alcoxy-peptoïdes étudiés.

Structure RX de l’homodimère Ac-(NOH)2-Pip. Liaisons H entre deux molécules arrangées en feuillet β antiparallèle. Les distances sont mesurées entre les atomes d’oxygène.
Les N-alcoxy-peptoïdes portant la chaîne O-(1-phényléthyle), (Orpe), ont été étudiés jusqu’à sept résidus de longueur, notamment par DC dans l’acétonitrile. Un effet coopératif est observé par accroissement de la longueur des oligomères, ce qui est un signe de structuration, mais une fois encore il est surprenant de constater que les spectres DC ressemblent à ceux des α-peptoïdes (Npe) [30], β-peptoïdes [40,41] ou encore α,β-peptoïdes [11].
Les N-alcoxyamides sont caractérisés par une pyramidalisation de l’azote, comme montré sur le N-méthoxy-N-méthylacétamide. L’optimisation de géométrie de ce modèle a ensuite servi à construire un modèle hexamèrique (NOspe)6 révélant, comme pour les N-Arylpeptoïdes une conformation de type PPII.
4 Contrôle de la conformation par des interactions locales
Les peptoïdes sont capables d’adopter des structures secondaires bien définies en solution mais l’isomérie cis/trans des jonctions amides est néanmoins un frein à la prédiction et au développement de structures tridimensionnelles homogènes [45]. La structuration des peptides et de nombreux oligoamides artificiels repose sur des liaisons hydrogène intramoléculaires. Le squelette des peptoïdes qui est dépourvu de NH d’amide, donneur de liaison hydrogène, n’est pas en mesure d’établir de telles interactions. Récemment, diverses stratégies ont été envisagées pour contrôler la géométrie des amides et de façon plus générale la flexibilité conformationnelle des peptoïdes. La possibilité de rétablir une liaison H entre un CO du squelette et une chaîne latérale donneuse de liaison H, de la même unité a été étudiée par le groupe de Blackwell, sur des peptoïdes modèles (Fig. 15) [46].

Exemples de chaînes latérales établissant une liaison H avec le squelette peptoïde.
L’influence de l’encombrement stérique des chaînes latérales sur le ratio cis/trans des amides a déjà été évoquée. Plus la différentiation stérique entre la chaîne latérale et le squelette est prononcée, plus l’équilibre rotamérique cis/trans est déplacé vers la forme cis. Cet effet a également été évalué à l’aide de petits modèles peptoïde [46b]. Deux types d’interactions électroniques locales ayant un effet sur l’équilibre cis/trans ont également été mis en évidence dans des peptoïdes modèles [46a,b]. Un transfert électronique n→π* entre carbonyles d’amides adjacents dans le squelette (CiOi→Ci+1Oi+1), uniquement possible lorsque le lien amide est trans, favorise cette géométrie au dépend de la conformation cis (Fig. 16a). Ce type de délocalisation électronique (n→π*Am), qui est par ailleurs impliqué dans la stabilisation des amides trans des hélices polyproline II [47], peut entrer en compétition avec un autre transfert électronique (n→π*Ar), lorsque la chaîne latérale est de type benzylique (Fig. 16b). L’interaction n→π*Ar, qui favorise la conformation cis correspond à un transfert électronique d’un carbonyle du squelette vers l’orbitale anti-liante du groupe aromatique de la chaîne latérale du résidu i+1.

Délocalisations électroniques locales dans les peptoïdes. a : délocalisation n→π*Am entre carbonyles adjacents ; b : délocalisation n→π*Ar entre un carbonyle et une chaîne aromatique.
Cette interaction est modulable puisqu’elle dépend du statut électronique de l’aromatique. Blackwell et al. ont évalué par RMN 1H dans différents solvants, la proportion de rotamères cis/trans de l’acétamide N-terminal des peptoïdes modèles A et B, portant notamment, des chaînes latérales phényléthyle diversement substituées sur l’aromatique (Fig. 17). Les groupes électroattracteurs sur l’aromatique (R = pNO2, F vs R = H, modèles A) déplacent l’équilibre vers le rotamère cis, alors que les groupes donneurs exercent l’effet inverse (R = O- vs R = H, modèles B). La chaîne cyclohexyléthyle (ch), non aromatique conduit à une diminution du rapport Kcis/trans par rapport à la chaîne pe. La chaîne pyridinum (pyr) très appauvrie en électrons, est celle qui donne la plus grande proportion de rotamère cis (Kcis/trans = 7,8 ; modèle B) dans cette étude. Les auteurs montrent également par expériences NOESY, que la conformation dans laquelle les groupes carbonyle et aromatique sont éclipsés est beaucoup plus peuplée dans le cas de la chaîne pyr que dans le cas de la chaîne pe, confirmant la contribution du transfert n→π*Ar dans la stabilisation de la conformation cis. Le groupe 1-cyanoéthyle (ce) proposé par Caumes et al. [48] stabilise également très fortement la conformation cis (Kcis/trans = 8,2). Une contribution n→π*CN n’est pas à exclure mais elle n’a pas encore été démontrée.

Étude des effets électroniques influençant l’interaction n→π*Ar. Les Kcis/trans sont mesurés dans CD3CN.
Le même groupe a également proposé la chaîne latérale de type triazolium (Fig. 18a) [48]. Elle est construite en deux étapes :
- • post-modification d’une chaîne latérale propargyle par cycloaddition de Huisgen catalysée au cuivre (CuAAC) ;
- • méthylation du triazole formé. L’aptitude du motif triazolium à déplacer l’équilibre rotamérique vers la conformation cis a également été évaluée à l’aide de peptoïdes modèles A.

Contrôle de la conformation par la chaîne triazolium. a : représentation de la chaîne triazolium et rapports Kcis/trans mesurés dans CD3CN ; b : conformation d’énergie minimale (B3LYP/6-31G + *) et interactions stabilisant la conformation cis de l’acétamide.
C’est aujourd’hui le meilleur inducteur d’amide cis avec en plus l’avantage unique de permettre la construction de chaînes latérales diversement substituées (groupe R). Les rotamères trans ne sont plus observés dans CDCl3 et la population d’amide cis est supérieure à 90 % dans CD3CN ou MeOD. Dans D2O, la proportion d’amide cis reste supérieure à 85 %, et fait remarquable, elle reste aussi élevée même en l’absence de substituant sur le carbone N-Cα. Une étude de la conformation par RMN, et DFT avec analyse Natural Bond Orbital (NBO) de la structure de plus basse énergie, a ensuite permis de montrer que les critères géométriques nécessaires à l’établissement d’une interaction n→π*Ar étaient satisfaits. Une liaison hydrogène entre le CH du triazolium et le carbonyle de la même unité permet une stabilisation supplémentaire de la conformation (Fig. 18b).
5 Conclusion
La famille des α-peptoïdes est apparue il y a maintenant 20 ans dans le contexte de la synthèse combinatoire de banques de peptidomimétiques. De nouvelles architectures de type peptoïde, résultant soit de modifications au niveau du squelette, soit de modifications portant sur la nature des chaînes latérales ont récemment enrichi cette famille. Les α-peptoïdes constituent une classe unique d’oligoamides foldamères capables de présenter des séquences d’amides « tout cis » (PPI), « tout trans » (PPII) ou encore une combinaison d’amides cis et trans comme dans la conformation « threaded loop ». Cette diversité de structuration repose en réalité sur une faible différence d’énergie libre entre les conformations cis et trans des amides N,N-disubstitués des peptoïdes, et conduit fréquemment à une hétérogénéité conformationnelle. Les travaux récents sur les α-peptoïdes se sont orientés vers la recherche de nouvelles chaînes latérales, capables par l’établissement d’interactions locales, de contrôler la géométrie cis/trans des amides, et de façon plus générale, de restreindre la flexibilité du squelette peptoïde. Par ailleurs, d’autres unités apparentées aux peptoïdes telles les N-hydroxy/alcoxy-glycines forment majoritairement des amides trans. Ces nouveaux outils devraient permettre d’atteindre dans un avenir proche de nouveaux foldamères caractérisés par des séquences contrôlées d’amides cis et trans. Dans le domaine des peptidomimétiques, ces nouveaux outils devraient également permettre de concevoir des ligands peptoïde possédant de meilleures affinités. L’étude de la conformation des β- et α,β-peptoïdes sera également grandement facilitée par l’emploi de chaînes latérales qui suppriment l’isomérie cis/trans.
Remerciements
Nous remercions toutes les personnes étroitement associées à nos travaux dans le domaine des peptoïdes, notamment les docteurs Thomas Hjelmgaard (KU-life, Danemark), Alison A. Edwards (Medway School of Pharmacy, Royaume-Uni) et Claude Didierjean (CRM2, France). Nous remercions également vivement Émiliana De Santis (doctorante, Medway School of Pharmacy, Royaume-Uni) pour sa contribution en dichroïsme circulaire.
1 (a) R.N. Zuckermann, D.A. Goff, S. Ng, K. Spear, B.O. Scott, A.C. Sigmund, R.A. Goldsmith, C.K. Marlowe, Y. Pei, L. Richter, R. Simon US005877278A 1999. (b) R.N. Zuckermann, D.A. Goff, S. Ng, K. Spear, B.O. Scott, A.C. Sigmund, R.A. Goldsmith, C.K. Marlowe, Y. Pei, L. Richter, R. Simon WO96/40202 1996. (c) R.N. Zuckermann, J.M. Kerr, S. Kent, W.H. Moos, R.J. Simon, D.A. Goff WO94/06451 1994.
2 Les azapeptoïdes rapportés par Kodadek et al. sont différents structuralement des hydrazino-aza, retro hydrazino-aza et N-azapeptoïdes rapportés par les groupes de Baudy-Floc’h et Le Grel. Voir par exemple : Cheguillaume et al. [15b] et Aubin et al. [15c].
3 Pour une structure RX d’un β-peptoïde cyclique : Roy et al. [42b].