

1 Introduction
Of the different chemical systems employed in the oxidation of drugs aiming to mimic biotransformation by cytochrome P450s or peroxidases, one of the most efficient associates a redox-active metalloporphyrin catalyst and a single oxygen atom donor such as sodium hypochlorite, potassium hydrogenpersulfate (KHSO5) or magnesium monoperphthalate (MMPP). These catalytic systems can indeed oxidize a variety of substrates either in aqueous or organic solution, and perform, amongst other transformations, the N-demethylation of tertiary amines [1–4].
SSR180575, a ligand of the peripheral benzodiazepine receptor, has been shown to improve functional recovery in induced neuropathies in the rat. Thus, it has been proposed as a treatment to promote neuronal survival and repair in the treatment of neurodegenerative diseases [5]. SSR180575 contains both N-methyl indole and N,N-dimethylamide functions, both being potential sites for N-demethylation by P450s, or by metalloporphyrin/oxygen donor (OD) systems. Thus, we have investigated the oxidation of SSR180575 using metalloporphyrin-catalyzed chemical systems, focusing on the relative reactivity of the N-methyl indole and the N,N-dimethylamide functions.
Several papers deal with the oxidative metabolism of N,N-dimethyl- amines or amides. N-demethylation of amines by P450s occurs by hydroxylation of the methyl group to a carbinolamine intermediate. The subsequent loss of a formaldehyde molecule affords the demethylated derivative. Demethylation of methylamides is less common, since the intermediate carbinolamide is more stable than a carbinolamine [6]. However, demethylation of dimethylformamide (DMF) to N-methylformamide (NMF) by the P450 2E1 isozyme has been considered as the first step of the process responsible for the hepatotoxicity of this industrial solvent [7–10]. This reaction occurs via initial hydroxylation of a methyl group, as assessed by a high intramolecular kinetic deuterium effect (KIE) either for the urinary metabolites of NMF in mice [9] or for metabolism of DMF by rat liver microsomes [8]. Intramolecular KIE values ranging from 3.6 to 6.9 were also reported for the demethylation of a series of dimethylamide derivatives. Such a high primary KIE is consistent with a mechanism involving hydrogen atom abstraction as the rate determining step, and suggest that a P450-catalyzed C-hydroxylation initiates the metabolism of such formamide derivatives [11]. Hydroxylation was also proposed as the first step of P450 oxidative demethylation of N-nitrosodimethylamine, responsible for the hepatocarcinogenicity of nitrosoalkylamines [12]. Hydroxylation of dialkylamide functions is also involved in the metabolism of drugs by P450, as an intermediate step during dealkylation [13]. However, N-hydroxymethylamide derivatives are considered to be unstable in protic or acidic conditions, and may thus be frequently overlooked in drug metabolism pathways [6]. The P450 metabolism of drugs containing both alkylamine and alkylamide functions involved dealkylation of the amine as a major pathway, and dealkylation of the amide as a minor – or not detectable pathway [14,15]. The oxidation of caffeine by P450 1A2 mainly produced the N3-demethylated metabolite, paraxanthine. The high KIE of this reaction (kH/kD = 7) strongly supports the formation of a N-hydroxymethyl intermediate which was not characterized [16].
In addition to their putative value for mimicking metabolic studies, chemical systems able to catalyze hydroxylation and demethylation of amide derivatives should allow an easy preparation of potentially reactive metabolites.
2 Results and discussion
SSR180575 (1, Fig. 1) contains both a dimethylamide function and an aromatic tertiary amine which can compete as sites for N-demethylation. Catalytic oxidation of SSR180575 was carried out using KHSO5 or MMPP, associated with different manganese porphyrin complexes (Fig. 2), in a biphasic dichloromethane/water medium. Benzyl dimethyl tetradecylammonium chloride (BDTAC) was added as a phase-transfer agent, with 4-t-butylpyridine as a cocatalyst [1], and the reaction was monitored by LC-MS. In the absence of metalloporphyrin, the conversion of 1 was in all cases below 5%.

Structure of SSR180575, 1.

Structures of the manganese complexes of tetramesitylporphyrin [Mn(TMP)Cl], tetrakis(3-chloro-2,4,6-trimethylphenyl)-β-octachloroporphyrin [Mn(Cl12TMP)Cl], and tetrakis(2,6-dichlorophenyl)porphyrin [Mn(TDCPP)Cl].
Under these conditions, SSR180575 was oxidized with variable efficacy and selectivity, giving mainly three products which coeluted in HPLC as a single peak having a shorter retention time than the substrate. We assumed that oxidation would leave the aromatic system unchanged, and should consequently not induce a significant change in the absorbance of the drug at 254 nm. For convenience, we considered that 1 and its oxidation products would have similar extinction coefficients. Calculated on this basis, the substrate conversion and selectivity in the main chromatographic peak after 30 min of reaction are reported in Table 1.
Conversion of SSR180575 by manganese porphyrin complexes associated with KHSO5 or MMPP after 30 min of reaction. Figures in parentheses stand for the selectivity of the HPLC peak at 7.4 min. Conversion and selectivity values are defined in experimental procedure section.
| Oxygen donor | Conversion of 1, % (selectivity at Rt 7.4 min, %) | ||
| Mn(TMP)Cl | Mn(TDCPP)Cl | Mn(Cl12TMP)Cl | |
| KHSO5a | 18 (60) | 40 (70) | 21 (56) |
| KHSO5b | 25 (51) | 45 (49) | nd |
| MMPPa | 45 (37) | 93 (50) | 43 (80) |
| MMPPb | 13 (80) | 100 (5) | 90 (33) |
a 3 mol equiv with respect to the substrate.
b 10 mol equiv with respect to the substrate.
For instance, the conversion of 1 catalyzed by Mn(TDCPP)Cl in the presence of 3 molar equivalents of KHSO5 was 40% after 30 min of reaction time, and the selectivity of the chromatographic peak eluted at 7.4 min was 70%. The mass spectrometry analysis of this chromatographic peak allowed detection of m/z 411.1, 393.1, 381.1, and 322.1 amu (with relative intensities 8, 100, 13, and 75, respectively), all of them having an isotopic pattern consistent with the presence of one chlorine atom (Fig. 3, panel b). The m/z 411.1 value should reasonably be assigned to the incorporation of an oxygen atom in the substrate (+16 amu), and the fragment at m/z = 393.1 amu (M-18) suggest the possible dehydration under the mass spectrometry conditions (ion 2′ or 3′, Fig. 4). Hydroxylation of the N-methylamide or N-methyl indole was then postulated (compounds 2 and 3, respectively, Fig. 4). The fragment peak with m/z = 322.1 was assigned to the loss of the dimethylamine chain (ion 2′′, Fig. 4). This fragmentation, with creation of a carbocation –CH2+ at C25, is not apparent in the spectrum of the substrate under the same conditions. So, we postulated that hydroxylation mainly occurred on the N-dimethylamide rather than on the N-methyl indole, thus weakening the amide bond. For the minor product with m/z = 381.1, N-demethylation of the substrate either on the amide- or indole functions may be postulated (compounds 4 and 5, respectively, Fig. 4). Significantly, when the quantity of KHSO5 was increased up to ten equivalents, the conversion of the substrate was hardly improved (45%), but the yield of the chromatographic peak at 7.4 min decreased to 49%, suggesting that over-oxidation may be occurring. A similar phenomenon was observed when 1 was oxidized by MMPP in the presence of Mn(Cl12TMP)Cl: conversion increased from 43 to 90% when the amount of MMPP was increased from 3 to 10 molar equivalents, but the selectivity decreased from 80 to 33%. The oxidation of 1 with 3 molar equivalents of MMPP, catalyzed by Mn(Cl12TMP)Cl, offered the best compromise between catalytic efficiency and selectivity. Using these conditions, after 30 min, conversion of 1 was 43% and the selectivity of the peak at 7.4 min was as high as 80%.
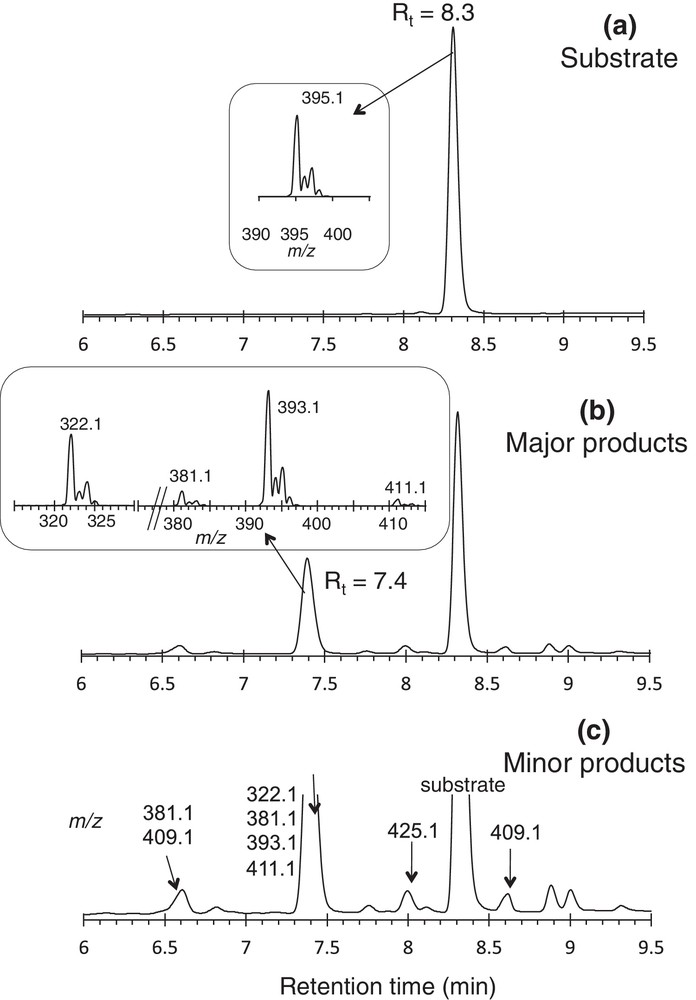
Oxidation of 1 by Mn(TDCPP)Cl/KHSO5. LC-MS analysis: UV detection at 254 nm at t = 0 (a) and 30 min (b). The ESI-MS spectra of the substrate and major oxidation products are depicted in inserts.

Hydroxylation of SSR180575 by Mn(Cl12TMP)Cl/MMPP. The mixture of compounds 2a, 2b, and 3 corresponds to the HPLC retention time = 7.4 min. Arrows indicate the most significant 1H NMR NOE correlations.
In order to further characterize the oxidation products, oxidation of 1 was performed on a larger scale with 3 mol equiv of MMPP in the presence of Mn(Cl12TMP)Cl. After 2 h, the reaction mixture was chromatographed on a silica gel column in order to isolate the compound mixture having an HPLC retention time of 7.4 min. The mixture thus obtained was analyzed by NMR, allowing characterization of products 2a, 2b, and 3 (see Fig. 4 for the structures). The 1H NMR data of compounds 2a, 2b, and 3 are reported in Table 2, for an easy comparison with those of the substrate 1. The molar proportions of 2a/2b/3 were 64/32/4 based on integration of the signals assigned to H2C25.
Selected 1H NMR chemical shifts for compounds 1, 2a, 2b, and 3 (500.0 MHz, DMSO-d6, 301 K). Underlined chemical shift values are those of –CH2OH functions.
| Compound | Cycle A | N-methyl indole | Cycle D | Amide chain | |||||||
| H9 | H10 | H12 | H14 | H16 H20 | H17 H19 | H18 | H23 | H24 | H25 | OH | |
| 1 | 7.89 | 7.41 | 7.96 | 4.29 | 7.60 | 7.53 | 7.44 | 3.19 | 2.87 | 4.24 | – |
| 2a | 7.98 | 7.39 | 7.98 | 4.31 | 7.60 | 7.53 | 7.44 | 4.96 | 2.89 | 4.38 | 6.25 |
| 2b | 7.90 | 7.42 | 7.98 | 4.31 | 7.60 | 7.53 | 7.44 | 4.73 | 3.19 | 4.25 | 5.85 |
| 3 | –a | 7.41 | 8.03 | 6.22 | –a | –a | –a | 3.20 | 2.87 | 3.98 | 6.68 |
a Overlapped with resonances of 2a and 2b.
The major product was the N-hydroxymethylamide derivative 2 existing as two conformers 2a/2b in a 2/1 ratio at 301 K (m/z = 411.1, MH+). The signals assigned to the hydroxyl group were detected at 6.25 and 5.85 ppm in 2a and 2b, respectively, and the HOCH2 resonance was detected at 4.96 and 4.73 ppm, respectively. The other signals were only slightly modified with respect to those of SSR180575 1. In the major conformer 2a, NOE correlations between (i) HOH2C23 and H9, (ii) HO and H9, and (iii) HOH2C23 and H2C25, confirmed the position of the N-hydroxymethylamide chain close to the indole cycle. In conformer 2b, significant NOE correlations were observed between (i) H3C24 and H9, (ii) H3C24 and H2C25, indicating that the methylamide was close to H9 and the hydroxymethyl chain orientated away from the polycyclic system. As a minor component of the mixture, compound 3, resulting from methyl hydroxylation of the N-methyl indole function, was also detected, with the hydroxyl proton at 6.68 ppm, and HOH2C14 at 6.22 ppm. Under these experimental conditions, the N-demethylated derivative (m/z = 381.1), which had been detected by LC-MS, was not isolated.
Beside the major HPLC peak with Rt 7.4 min, several minor peaks were detected, each of them accounting for less than 1% of the observed selectivity (Fig. 2, panel b). In some cases, isotopic patterns were characteristic for compounds containing two chlorine atoms, possibly formed by a non-catalytic chlorination involving dichloromethane.
In conclusion, the catalytic oxidation of SSR180575 provided the N-hydroxymethyl-N-methylamide derivatives 2a/2b as the main oxidation products. By contrast, only a very small amount of N-monodemethylated product was detected by LC-MS, and we failed to detect any didemethylated product (expected m/z = 367.1 amu). In addition, no product due to a one-electron oxidation of the indole ring could be detected. This result is consistent with a rather high redox potential for one-electron oxidation of the indole moiety due to (i) the acidic conditions [17], and (ii) the substitution of the indole by the electron-withdrawing chlorine atom at position 6. Under similar oxidation conditions using a manganese porphyrin complex associated with KHSO5, hydroxylation of the N,N-dimethylamide function of another drug was also obtained, despite in a lower yield than with SSR 180575 as a substrate [unpublished data]. This suggests that this catalytic system should be an efficient P-450 mimic for a variety of substrates, and may help for an useful reaction in organic synthesis.
Few reports deal with the chemically catalyzed demethylation of amides. The oxidation of N-methyl lactams catalyzed by iron tetraphenylporphyrin [Fe(TPP)] associated with t-butylhydroperoxide gave rise to ring oxidation with an overwhelming preference with respect to N-demethylation. By contrast, when oxidation was carried out by liver microsomes, N-demethylation is preferred to ring oxidation [18]. These results suggest a radical reaction of the Fe(TPP)/t-BuOOH system, as expected when using a hydroperoxide as oxidant [19], and indicate the possibility of modulating the oxidation properties of synthetic catalytic systems. Our previous oxidation of SR48117, an antagonist of vasopressine V1a, resulted in mono- and didemethylation of a dimethylamide function, but we failed to isolate a hydroxymethylamide intermediate, the putative P450 metabolite [4].
3 Conclusion
The catalytic oxidation of SSR180575 has several original features:
- • a P450-type reactivity leading to hydroxylation of a methyl group of the N-dimethylamide chain in high yield. In contrast to the case of DMF and NMF, the hydroxylated derivative of SSR180575 does not have a formamide structure, but an acetamide-type side chain which precludes the formation of an isocyanate intermediate and the related toxicity;
- • higher oxidation sensitivity of the N-dimethylamide with respect to the N-methyl indole;
- • formation of a carbinolamide derivative which is stable enough to be isolated, even in acidic medium (pH 2 in the reaction mixture) and in the presence of protic solvents (2 vol% methanol). N-hydroxymethylamide derivatives are usually considered unstable in protic or acidic conditions [6].
4 Experimental procedures
4.1 Chemicals
Metalloporphyrin complexes were synthesized according to a published procedure [20]. SSR180575 was synthesized by Sanofi Research & Development (Chilly-Mazarin, France). All commercially available products and solvents were purchased from Sigma-Aldrich-Fluka, and used as received. The oxidants used were potassium monopersulfate (as a triple salt 2KHSO5·KHSO4·K2SO4), and magnesium monoperoxyphtalate hexahydrate (MMPP).
4.2 Analyses
1H- and 13C-NMR spectra were recorded in DMSO-d6, at 301 K, on a Bruker Avance 500 spectrometer (500.0 MHz for 1H, and 125.7 MHz for 13C), using tetramethylsilane (TMS) as the external standard. LC-MS analyses were performed using an Agilent 6140 triple-quadrupole machine equipped with an electrospray source (positive mode), and a Kromasil C18 column (3.5 μm, 150 × 3 mm). Eluent A was water/acetonitrile/methanesulfonic acid, 1000/25/1, v/v/v, and eluent B was water/acetonitrile/methanesulfonic acid, 25/1000/1, v/v/v. The elution gradient was linear from A/B = 100/0 to A/B = 0/100 in 7 min, followed by a further 8 min at A/B = 0/100. The flow rate was 0.6 mL.min−1, and UV detection was carried out at 254 nm. The mass spectrometry analyses were performed in scan mode (m/z range = 100–1300 amu). Under these conditions, the retention time of SSR180575 1 was 8.3 min. Conversion of the substrate at time t was calculated as [(abs 1 at t0 – abs 1 at t)/abs 1 at t0] × 100. Selectivity of an HPLC peak having a retention time Rt was calculated as [abs at Rt/(abs 1 at t0 – abs 1 at t)] × 100, where abs stands for absorbance at 254 nm.
4.3 General conditions for oxidation reactions
The drug (SSR180575, 20 μmol, 7.9 mg) was dissolved in 0.4 mL of dichoromethane containing the metalloporphyrin [0.4 μmol, corresponding to 0.34 mg, 0.39 mg, or 0.51 mg of Mn(TMP)Cl, Mn(TDCPP)Cl, or Mn(Cl12TMP)Cl, respectively], 4-t-butylpyridine (10 μmol, 1.5 μL), and benzyldimethyltetradecylammonium chloride (BDTAC, 15 μmol, 5.5 mg). The oxidant (60 μmol corresponding to 18.4 mg or 29.7 mg of KHSO5 or MMPP, respectively), dissolved in 0.8 mL of water, was then added. The mixture was kept at 18–20 °C, under magnetic stirring.
Aliquots of the organic phase (4 μL) were withdrawn before addition of the oxidant, and after every 30 min of reaction, diluted in a mixture H2O/CH3CN/CH3SO3H, 50/50/0.1, v/v/v, and analyzed by LC-MS.
4.4 Purification and characterization of compounds 2a, 2b, and 3
The above-mentioned general conditions for oxidation of 1 by Mn(Cl12TMP)Cl/MMPP were scaled-up 12 fold (100 mg of 1). After 2 h of reaction, the organic phase was recovered. The aqueous phase was washed twice with dichloromethane. The organic extracts were combined, washed with water (10 mL), then brine (10 mL), dried over MgSO4, filtered, and evaporated to dryness. The residue was purified through a silica gel column, with a mixture dichoromethane/methanol as eluent (gradient from 0 to 2 vol% of CH3OH). The fraction containing 2 and 3 was collected and evaporated. The products were then dissolved in a minimum amount of dichloromethane, and precipitated by addition of diethyl ether, affording 30 mg of the mixture of products 2a, 2b, and 3 in proportion 64/32/4 (overall yield = 29%).
NMR characterization was carried out directly on the mixture 2a/2b/3. For clarity, NMR characterizations are described separately. 1H resonances are collected in Table 2. 13C-NMR for 2a δ 169.4 (OC21), 154.9 (OC3), 142.1 (C15), 141.5 (C13), 141.4 (C6), 132.4 (ClC11), 131.0 (NC2), 129.0 (C17 and C19), 128.1 (C18), 126.8 (C16 and C20), 124.1 (C9), 122.8 (C10), 119.1 (C8), 117.6 (C7), 111.9 (C12), 73.1 (HOC23N), 38.8 (C25), 33.0 (NC24), 32.2 (N–C14).
13C-NMR for 2b δ 169.0 (OC21), 154.9 (OC3), 142.1 (C15), 141.5 (C13), 141.5 (C6), 132.4 (ClC11), 131.1 (NC2), 129.0 (C17 and C19), 128.2 (C18), 126.7 (C16 and C20), 124.3 (C9), 122.9 (C10), 119.0 (C8), 117.6 (C7), 111.9 (C12), 69.8 (HOC24N), 39.3 (C25), 33.9 (NC23), 32.2 (NC14).
13C-NMR for 3 (selected data) δ 168.7 (OC21), 140.8 (C6), 132.4 (ClC11), 123.2 (C10), 119.7 (C8), 117.2 (C7), 112.7 (C12), 67.6 (HOC14N), 41.2 (C25), 37.5 (NC23), 35.5 (NC24).
Acknowledgements
This work was supported by Sanofi Recherche & Développement and the CNRS.