

1 Introduction
Phosphorus ylides are important reagents in organic chemistry, especially in the synthesis of naturally occurring products with biological and pharmacological activities [1]. The utility of metalated phosphorus ylides in synthetic chemistry has been well documented [2–4]. Ylides play in coordination chemistry other roles aside from its coordination to the metal [5]. Juxtaposition of the keto group and of the carbanion in the phosphorus ylides allows resonance delocalization of the ylidic electron density, providing additional stabilization to the ylide species. This so-called α-stabilization provides ylides with the potential to act as an ambidentate ligand and thus bond to a metal centre through the either carbon (B) or oxygen (A1 and A2) (Scheme 1). Although many bonding modes are possible for keto ylides [6], coordination through carbon is more predominant and observed with soft metal ions, e.g., Pd(II) [7], Pt(II) [7h,8], Ag(I) [9], Hg(II) [9d,10], Au(I) [11], Cd(II) [7i,9d,12], whereas O-coordination dominates when the metals involved are hard, e.g., Ti(IV), Zr(IV), and Hf(IV) [13].
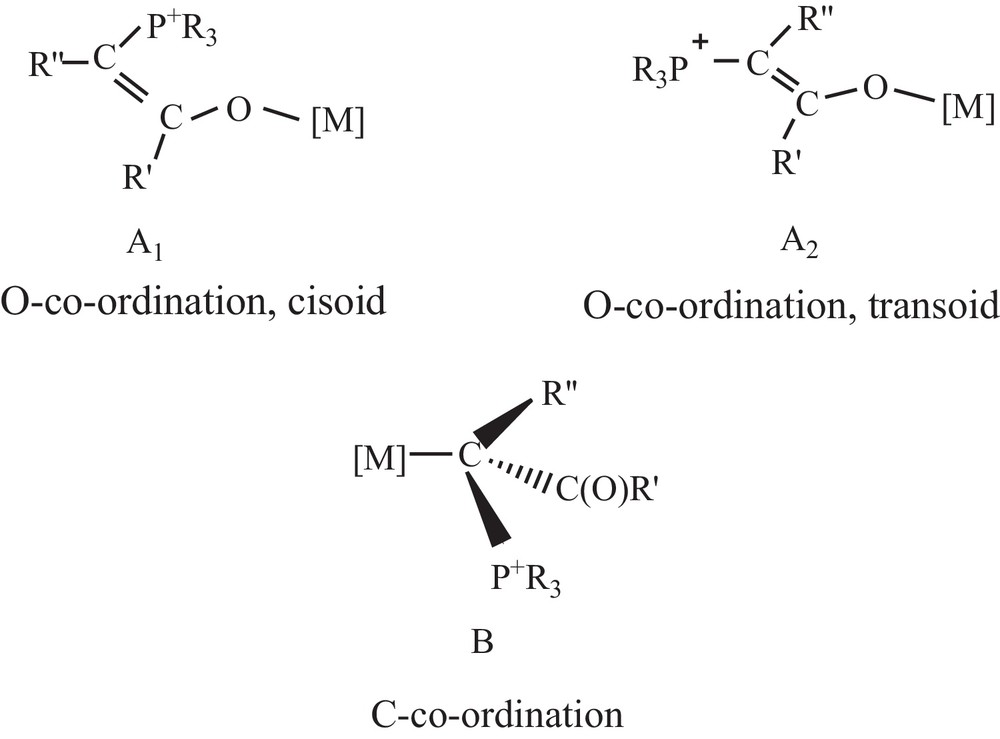
The possible bonding modes of an α-keto stabilized ylide to the metal center.
Synthesis of complexes derived from phosphorus ylides and Hg(II) halides was initiated in 1965 by Nesmeyanov et al. [14] In 1975, Weleski et al. [15] proposed a symmetric halide-bridged dimeric structure for Hg(II) halide complexes, whereas Kalyanasundari et al. [10a] reported an asymmetric halide-bridged dimeric structure in 1995. In 1985, Sanehi et al. [16] reported a mononuclear Hg(II) complex of phosphorus ylides without any structural characterization. We have recently worked on the synthesis of mononuclear [9d,10b], binuclear [17–20] complexes derived from mercury (II) salts and phosphorus ylides.
Based on the above backgrounds, we have focused on the synthesis, structure, and properties of coordination mononuclear Hg (II) and Cd (II) complexes with ambidentate phosphorus ylide as a ligand, and DMSO has been successfully utilized for the preparation of a series of complexes. In this study, we describe the preparation, structural and spectroscopic characterization of Hg(II) and Cd(II) complexes with the title ylide. Herein the structure of a new mononuclear mercury complex was reported and the single-crystal X-ray diffraction of 6 demonstrates the C-coordination of the ylide and O-coordination of the DMSO to the metal center.
2 Experimental
2.1 Materials and measurements
Methanol and dichloromethane were distilled over magnesium powder and diethyl ether (Et2O) over a mixture of sodium and benzophenone just before use. All other solvents were reagent grade and used without further purifications. The ligand L was prepared and characterized according to the published procedure [17]. Melting points were measured on a SMPI apparatus. Elemental analyses for C, H and N were performed using a PerkinElmer 2400 series analyzer. Fourier transform IR spectra were recorded on a Shimadzu 435-U-04 spectrophotometer and samples were prepared as KBr pellets. 1H, 13C and 31P NMR spectra were recorded on a 300 MHz Bruker and 90 MHz Jeol spectrometer in DMSO-d6 or CDCl3 as a solvent at 25 °C. Chemical shifts (ppm) are reported according to internal TMS and external 85% phosphoric acid. Coupling constants are given in Hz.
2.2 Crystallographic data collection and structure determination
The single-crystal X-ray diffraction analyses of suitable crystal of 6 was performed on a STOE IPDS-II diffractometer at 298 K, using the graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data collection was performed using the ω-scan technique and using the STOE X-AREA software package [21]. The crystal structure was solved by direct methods and refined by full-matrix least-squares on F2 by SHELX [22] and using the X-STEP32 crystallographic software package [23]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions using a riding mode with fixed thermal parameters.
2.3 Synthesis and characterization of compounds
2.3.1 Ph3PCHCOC6H4Me (L)
Selected IR data (KBr, ν/cm−1), 1582 (νCO) and 895 (νP+–C−). 1H NMR (CDCl3, ppm): δ = 2.38 (s, 3H, CH3), 4.32 (d, 1H, 2JPH = 23.29 Hz, CH), 7.1–8.1 (m, 19H, arom). 31P NMR (CDCl3, ppm): δ = 12.98. 13C NMR (CDCl3, ppm): δ = 21.48 (s, CH3), 45.63 (d, 1JPC = 70.42 Hz, CH), 115.4 (d, 1JPC = 91.43 Hz, PPh3 (i)), 131.7 (d, 3JPC = 12.36 Hz, PPh3 (m)), 133.2 (d, 2JPC = 8.07 Hz, PPh3 (o)), 142.31 (s, PPh3 (p)), 145.50 (s, COPh (i)), 126.19 (s, COPh (m)), 132.03 (s, COPh (o)), 128.7 (s, COPh (p)), 191.34 (s, CO).
2.3.2 [HgCl(μ-Cl){CH(PPh3)C(O)C6H4Me}]2 (1)
General procedure: To HgCl2 (0.172 g, 0.63 mmol) dissolved in 5 mL of dried methanol was added L (0.250 g, 0.63 mmol) at room temperature. The mixture was stirred for 4 h. The white solid product was filtered, washed with diethyl ether and dried under reduced pressure. Yield: (84%); mp 208–210 °C; Selected IR data (KBr, ν/cm−1), 1632 (νCO) and 817 (νP+–C−). 1H NMR (DMSO-d6, ppm): δ = 2.35 (s, 3H, CH3), 5.42 (d, 1H, 2JPH = 3.85 Hz, CH), 6.94–8.12 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 24.60. 13C NMR (DMSO-d6, ppm): δ = 20.89 (s, CH3), 46.11 (d, 1JPC = 70.42 Hz, CH), 122.91 (d, 1JPC = 89.99 Hz, PPh3 (i)), 129.21 (d, 3JPC = 12.36 Hz, PPh3 (m)), .17 (d, 2JPC = 9.77 Hz, PPh3 (o)), 134.33 (d, 4JPC = 9.84 Hz, PPh3 (p)), 142.46 (s, COPh (i)), 128.19 (s, COPh (m)), 133.38 (s, COPh (o)), 128.54 (s, COPh (p)), 190.87 (s, CO). Anal. calcd. (%) for C54H46Cl4Hg2O2P2 (1332): C, 48.69; H, 3.48. Found (%): C, 48.45, H, 3.54.
2.3.3 [HgBr(μ-Br){CH(PPh3)C(O)C6H4Me}]2 (2)
Yield: (85%); mp 197–199 °C. Selected IR data (KBr, ν/cm−1), 1630 (νCO) and 814 (νP+-C−). 1H NMR (DMSO-d6, ppm): δ = 2.39 (s, 3H, CH3), 5.52 (br, 1H, CH), 7.04–8.24 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 23.46. 13C NMR (DMSO-d6, ppm): δ = 21.06 (s, CH3), 47.13 (d, 1JPC = 73.95 Hz, CH), 123.01 (d, 1JPC = 89.52 Hz, PPh3 (i)), 129.27 (d, 3JPC = 12.43 Hz, PPh3 (m)), 133.23 (d, 2JPC = 9.41 Hz, PPh3 (o)), 133.44 (s, PPh3 (p)), 142.41 (s, COPh (i)), 126.57 (s, COPh (m)), 131.97 (s, COPh (o)), 128.64 (s, COPh (p)), 190.75 (s, CO). Anal. calcd. (%) for C54H46Br4Hg2O2P2 (1510): C, 42.96; H, 3.07. Found (%): C, 43.21, H, 3.11.
2.3.4 [HgI(μ-I){CH(PPh3)C(O)C6H4Me}]2 (3)
Yield: (81%); mp 186–189 °C. Selected IR data (KBr, ν/cm−1), 1621 (νCO) and 797 (νP+–C−). 1H NMR (DMSO-d6, ppm): δ = 2.34 (s, 3H, CH3), 5.25 (d, 1H, 2JPH = 8.44 Hz, CH), 7.07–8.12 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 21.51. 13C NMR (DMSO-d6, ppm): δ = 20.83 (s, CH3), 44.23 (d, 1JPC = 72.49 Hz, CH), 123.61 (d, 1JPC = 87.65 Hz, PPh3 (i)), 128.93 (d, 3JPC = 12.57 Hz, PPh3 (m)), 132.86 (d, 2JPC = 9.19 Hz, PPh3 (o)), 135.07 (s, PPh3 (p)), 141.34 (s, COPh (i)), 127.58 (s, COPh (m)), 132.86 (d, 4JPC = 9.19 Hz, COPh (o)), 128.25 (s, COPh (p)), 189.40 (d, 2JPC = 15.97 Hz, CO). Anal. calcd. (%) for C54H46I4Hg2O2P2 (1698): C, 38.20; H, 2.73. Found (%): C, 38.37, H, 2.67.
2.3.5 [CdCl(μ-Cl){CH(PPh3)C(O)C6H4Me}]2 (4)
To CdCl2. H2O (0.150 g, 0.38 mmol) dissolved in 5 mL of dried methanol was added L (0.077 g, 0.38 mmol) at room temperature. The mixture was stirred for 12 h. The white solid product was filtered, washed with dietyl ether and dried under reduced pressure. Yield: (64%); mp 197–200 °C. Selected IR data (KBr, ν/cm−1), 1653 (νCO) and 739 (νP+–C−). 1H NMR (DMSO-d6, ppm): δ = 2.30 (s, 3H, CH3), 4.40 (d, 1H, 2JPH = 27.68 Hz, CH), 6.85–7.78 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 16.35. 13C NMR (DMSO-d6, ppm): δ = 20.77 (s, CH3), 44.56 (d, 1JPC = 72.56 Hz, CH), 121.30 (d, 1JPC = 90.31 Hz, PPh3 (i)), 128.83 (d, 3JPC = 12.07 Hz, PPh3 (m)), .66 (d, 2JPC = 10.36 Hz, PPh3 (o)), 132134.08 (s, PPh3 (p)), 153.23 (s, COPh (i)), 121.94 (s, COPh (m)), 126.60 (s, COPh (o)), 128.57 (s, COPh (p)), 183.66 (d, 2JPC = 2.43 Hz, CO). Anal. calcd. (%) for C54H46Cd2Cl4O2P2 (1156): C, 56.13; H, 4.01. Found (%): C, 56.22; H, 4.03.
2.3.6 [HgCl2{CH(PPh3)C(O)C6H4Me}(OSMe2)] (5)
General procedure: 0.150 g (0.1 mmol) of binuclear complex 1 was dissolved in DMSO (2 ml). The pale yellow crystals formed by the slow evaporation of the solvent over several days. Yield: (82%); Decomp. 183 °C. Selected IR data (KBr, ν/cm−1), 1635 (νCO) and 825 (νP+–C−), 1H NMR (DMSO-d6, ppm): δ = 2.41 (s, 3H, CH3), 5.36 (br, 1H, CH), 7.24–8.13 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 22.03. 13C NMR (DMSO-d6, ppm): δ = 22.25 (s, CH3), 47.89 (d, 1JPC = 74.14 Hz, CH), 122.21 (d, 1JPC = 90.09 Hz, PPh3 (i)), 130.14(d, 3JPC = 11.64 Hz, PPh3 (m)), 133.72 (d, 2JPC = 10.0 Hz, PPh3 (o)), 133.93 (s, PPh3 (p)), 143.21 (s, COPh (i)), 127.64 (s, COPh (m)), 132.0 (s, COPh (o)), 128.19 (s, COPh (p)), 189.58 (s, CO). Anal. calcd. (%) for C29H29HgCl2O2PS (744): C, 46.81; H, 3.93. Found (%): C, 46.72; H, 3.80.
2.3.7 [HgBr2{CH(PPh3)C(O)C6H4Me}(OSMe2)] (6)
Yield: (95%); Decomp. 196 °C. Selected IR data (KBr, ν/cm−1), 1628 (νCO) and 819 (νP+–C−). 1H NMR (DMSO-d6, ppm): δ = 2.42 (s, 3H, CH3), 5.65 (br, 1H, CH), 7.42–8.30 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 21.65. 13C NMR (DMSO-d6, ppm): δ = 21.06 (s, CH3), 47.13 (d, 1JPC = 73.95 Hz, CH), 123.01 (d, 1JPC = 89.52 Hz, PPh3 (i)), 129.27 (d, 3JPC = 12.43 Hz, PPh3 (m)), 133.23 (d, 2JPC = 9.41 Hz, PPh3 (o)), 133.44 (s, PPh3 (p)), 142.41 (s, COPh (i)), 126.57 (s, COPh (m)), 131.97 (s, COPh (o)), 128.64 (s, COPh (p)), 190.75 (s, CO). Anal. calcd. (%) for C29H29HgBr2O2PS (833): C, 41.82; H, 3.5. Found (%): C, 42.01; H, 3.4.
2.3.8 [HgI2{CH(PPh3)C(O)C6H4Me}(OSMe2)] (7)
Yield: (71%); Decomp. 224 °C. Selected IR data (KBr, ν/cm−1), 1628 (νCO) and 832 (νP+-C−). 1H NMR (DMSO-d6, ppm): δ = 2.39 (s, 3H, CH3), 5.19 (br, 1H, CH), 7.35–8.23 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 21.15. 13C NMR (DMSO-d6, ppm): δ = 21.0 (s, CH3), 48.58 (d, 1JPC = 72.85 Hz, CH), 123.29 (d, 1JPC = 90.68 Hz, PPh3 (i)), 129.84 (d, 3JPC = 14.01 Hz, PPh3 (m)), 134.28 (d, 2JPC = 11.51 Hz, PPh3 (o)), 134.72 (s, PPh3 (p)), 142.39 (s, COPh (i)), 127.18 (s, COPh (m)), 132.65 (s, COPh (o)), 129.46 (s, COPh (p)), 192.11 (s, CO). Anal. calcd. (%) for C29H29HgI2O2PS (927): C, 35.57; H, 3.15. Found (%): C, 35.12; H, 3.11.
2.3.9 [CdCl2{CH(PPh3)C(O)C6H4Me}(OSMe2)] (8)
Yield: (65%); Decomp. 183–185 °C. Selected IR data (KBr, ν/cm−1), 1635 (νCO) and 828 (νP+-C−). 1H NMR (DMSO-d6, ppm): δ = 2.12 (s, 3H, CH3), 5.0 (br, 1H, CH), 7.33–8.12 (m, 19H, arom). 31P NMR (DMSO-d6, ppm): δ = 23.12. 13C NMR (DMSO-d6, ppm): δ = 19.68 (s, CH3), 49.32 (d, 1JPC = 75.61 Hz, CH), 124.21 (d, 1JPC = 92.59 Hz, PPh3 (i)), 130.24 (d, 3JPC = 15.14 Hz, PPh3 (m)), 134.18 (d, 2JPC = 13.84 Hz, PPh3 (o)), 135.62 (s, PPh3 (p)), 143.08 (s, COPh (i)), 127.84 (s, COPh (m)), 133.12 (s, COPh (o)), 129.82 (s, COPh (p)), 187.13 (s, CO). Anal. calcd. (%) for C29H29CdCl2O2PS (656): C, 53.10; H, 4.46. Found (%): C, 52.41; H, 4.44.
3 Results and discussion
3.1 Chemistry
In this study, four binuclear complexes and four mononuclear complexes with the ambidentate phosphorus ylide (p-methylbenzoyl)methylene triphenylphosphorane were synthesized and investigated by physicochemical techniques. Reaction of L with MX2 (M = Hg; X = Cl, Br and I and M = Cd; X = Cl) in methanol (1:1) yielded the binuclear complexes (Scheme 2) [10a,17]. The bridge-splitting reaction of binuclear complexes [MX(μ-X){CH(PPh3)C(O)C6H4Me}]2 by DMSO yields the mononuclear complexes [MX2{CH(PPh3)C(O)C6H4Me}(OSMe2)] (Scheme 2) [13].
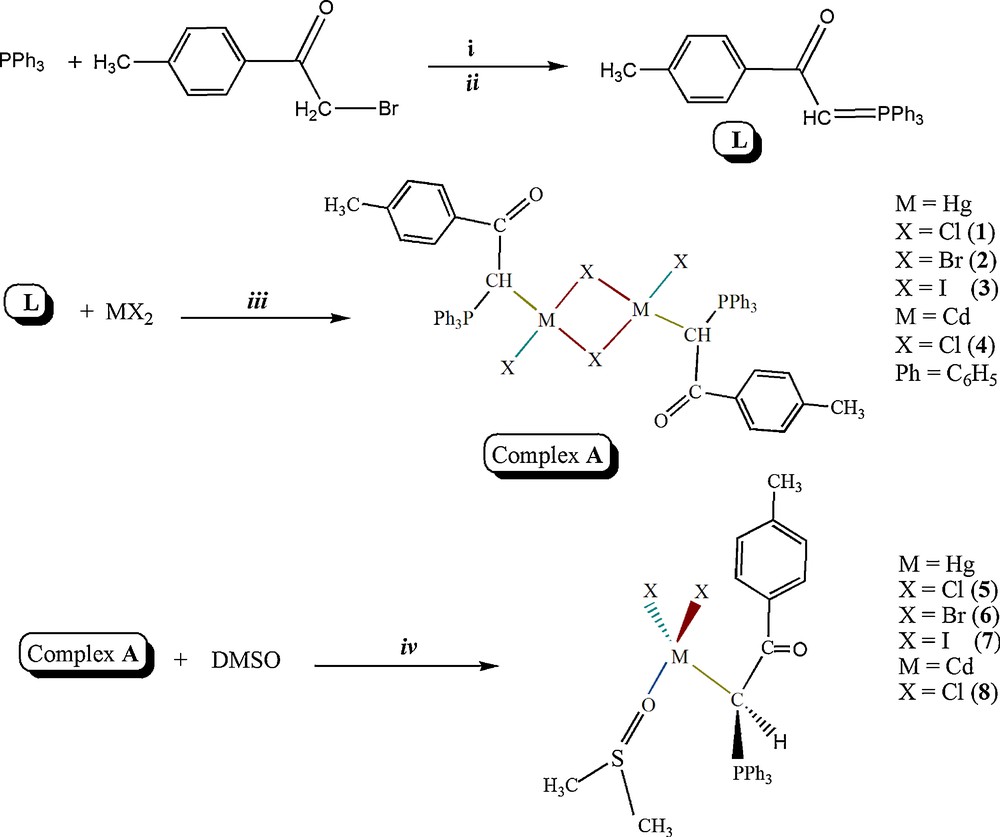
Synthetic route for the preparation of Hg(II) and Cd(II) complexes. Reagents and conditions: i: in acetone, using dry nitrogen atmosphere, 10 h at 25 °C; ii: in an aqueous solution of NaOH (0.5 mol), 2 h in 40 °C; iii: in methanol, for binuclear Hg complexes, 4 h at 25 °C; for binuclear Cd complexes, 12 h at 25 °C; iv: in dimethyl solfoxide, 3 days at 25 °C.
3.2 IR studies
The ν (CO) band, which is sensitive to complexation, occurs at 1582, in the parent ylides (L) [17–20,23]. Coordination of ylide through the carbon atom causes an increase in the ν (CO) band, whereas for O-coordination a lowering of the ν (CO) band is expected [10b]. Thus the IR absorption bands for the complexes at higher frequencies indicate that C-coordination has occurred. The ν(PC) band frequencies, which are also diagnostic of the coordination mode, occur at 895 in the parent ylides (L), and are shifted to lower frequencies for the complexes, suggesting some removal of the electron density of the PC bonds [24]. It should be noted that there is no significant difference in the IR absorption band between binuclear and related mononuclear complexes.
3.3 NMR studies
In the 1H NMR spectra, methinic protons exhibit doublet or broad doublet signals. Similar behavior was observed earlier in the case of ylide complexes of platinum(II) chloride [25]. The expected downfield shifts of 31P and 1H signals for the PCH group upon complexation in the case of C-coordination were observed in their corresponding spectra. It must be noted that O-coordination of the ylide generally leads to the formation of cisoid and transoid isomers, giving rise to two different signals in 31P and 1H NMR (Scheme 1) [7h]. The downfield shifts of the CH proton upon coordination in the 1HNMR spectra show the coordination of the ylides through methinic carbon atoms. The proton-decoupled 31P NMR spectra show only one sharp singlet between δ 16.2–24.6 ppm for the complexes of Hg(II) halides and CdCl2.H2O. Chemical shift values for the complexes appear to be downfield by about δ 2–10 ppm with respect to the parent ylide (δ 14.10 ppm for L), indicating that coordination of the ylide has occurred.
The most interesting aspect of the 13C NMR spectra of the complexes is the up-field shift of the signals due to the ylidic carbon. Similar up-field shifts of 2–3 ppm with reference to the parent ylide were also observed in the case of the mercury cyclopentadienyl complex [26] and in our synthesized Hg(II) complexes [9d,10b]. The 13C shifts of the CO group in the complexes are lower than noted for the same carbon in the parent ylide, indicating much lower shielding of the carbon of the CO group in these complexes.
3.4 X-ray structure analysis
Table 1 provides the crystallographic results and refinement information for complex 6. The molecular structure is shown in Fig. 1. Pertinent bond distances and angles for 6 are given in Table 2. The fractional atomic coordinates and equivalent isotropic displacement coefficients (Ueq) for the non-hydrogen atoms of the complex are shown in the supplementary material. The Hg(II) centre in complex 6 is coordinated by one carbon from ylide, one oxygen of coordinated DMSO and two bromo atoms in a distorted tetrahedral geometry. The two different Hg–Br distances in 6 (2.6166(10) and 2.5204(9) Å) are less than those found in the mononuclear complex of [HgBr2(PPh3)2] (2.633(6) and 2.626(8) Å) [27], indicating relatively strong HgBr bonds in 6. The difference between two distances in these complexes might be arising from steric effects of the large ylidic groups. The angles around mercury vary from 88.3(3) to 138.80(17), indicating a much distorted tetrahedral environment. This distortion must be due to the higher s character of the sp3 hybrid mercury orbitals involved in the above bonds and the steric effects of phosphine group causing the CHBr angle to be larger. In complex 6, a medium intermolecular interactions, including CH (25) Phenyl….Br (1) (3.044 Å) (symmetry operations used: 1.5–x, −½+y, ½–z), determine the structural assembly in this compound (Fig. 2).
Crystal data and refinement details for 6.
| Identification code | 6 |
| Empirical formula | C29H29Br2Hg1O2P1S1 |
| Formula weight | 832.95 |
| Temperature | 298(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P2/1n |
| Unit cell dimensions | a = 16.7187(16) Å |
| b = 10.7949(6) Å | |
| c = 18.6553(18) Å | |
| β = 115.677(7)° | |
| Volume (Å3) | 3034.4(4) |
| Z, calculated density (mg/m3) | 4,1.823 |
| Absorption coefficient (mm−1) | 7.851 |
| F(000) | 1600 |
| Crystal size (mm) | 0.30 × 0.15 × 0.10 mm |
| θ, range for data collection (°) | 2.17–29.25 |
| Limiting indices | −18 ≤ h ≤ 22 |
| −12 ≤ k ≤ 14 | |
| −25 ≤ l ≤ 25 | |
| Reflections collected/unique | 21,289/8119 [R(int) = 0.0919] |
| Completeness | 98.4% |
| Absorption correction | Numerical |
| Max. and min. transmission | 0.450 and 0.250 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 8119/0/325 |
| Goodness-of-fit on F2 | 0.899 |
| Final R indices [I > 2 σ(I)] | R1 = 0.0543, wR2 = 0.1073 |
| R indices (all data) | R1 = 0.1165, wR2 = 0.1254 |
| Largest diff. peak and hole (eÅ3) | 1.818 and −0.945 |
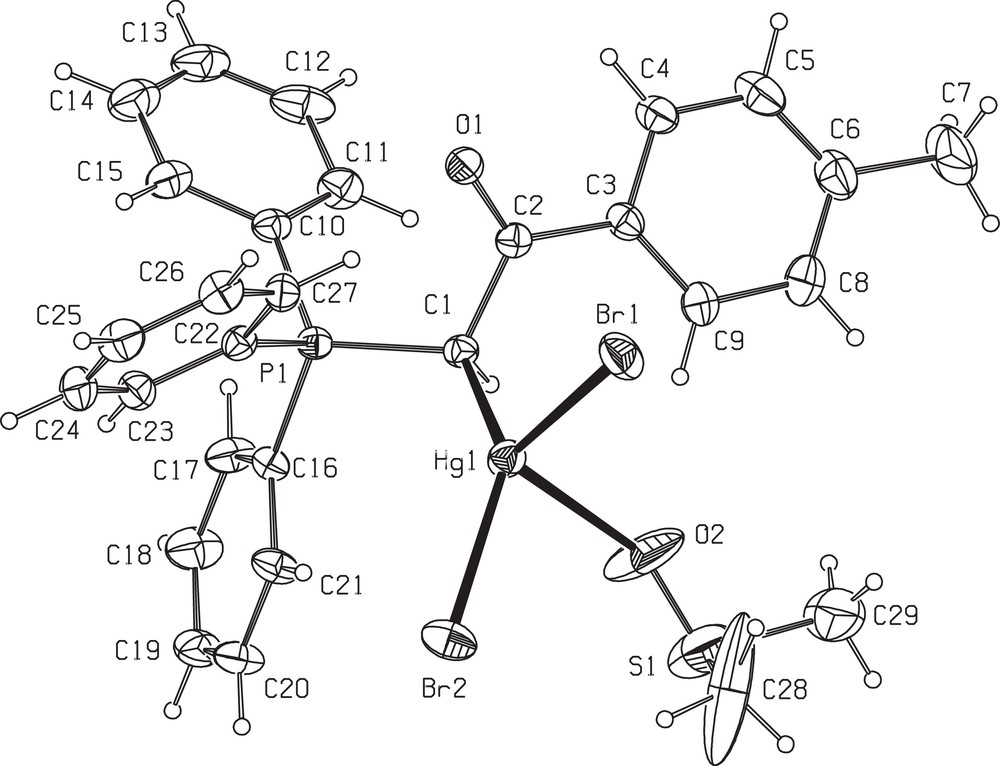
ORTEP view of the asymmetric unit of 6 (30% probability level) showing the numbering scheme.
Selected bond lengths (Å) and angles (°) for 6a.
| Bond lengths | |
| Hg(1)C(1) | 2.221 (7) |
| Hg(1)Br(1) | 2.6166 (10) |
| Hg(1)Br(2) | 2.5204 (9) |
| Hg(1)O(2) | 2.540 (7) |
| P(1)C(1) | 1.788 (7) |
| O(1)C(2) | 1.230 (8) |
| C(1)C(2) | 1.498 (10) |
| O(2)S(1) | 1.412 (8) |
| Bond angles | |
| Br(2)Hg(1)Br(1) | 106.33 (3) |
| C(1)Hg(1)Br(2) | 138.80 (17) |
| C(1)Hg(1)Br(1) | 113.13 (17) |
| C(1)Hg(1)O(2) | 88.3 (3) |
| O(2)Hg(1)Br(2) | 91.3 (2) |
| O(2)Hg(1)Br(1) | 106.9 (4) |
| C(2)C(1)P(1) | 112.8 (5) |
| C(2)C(1)Hg(1) | 109.3 (4) |
| P(1)C(1)Hg(1) | 110.5 (3) |
a See Fig. 1 for the atom numbering.
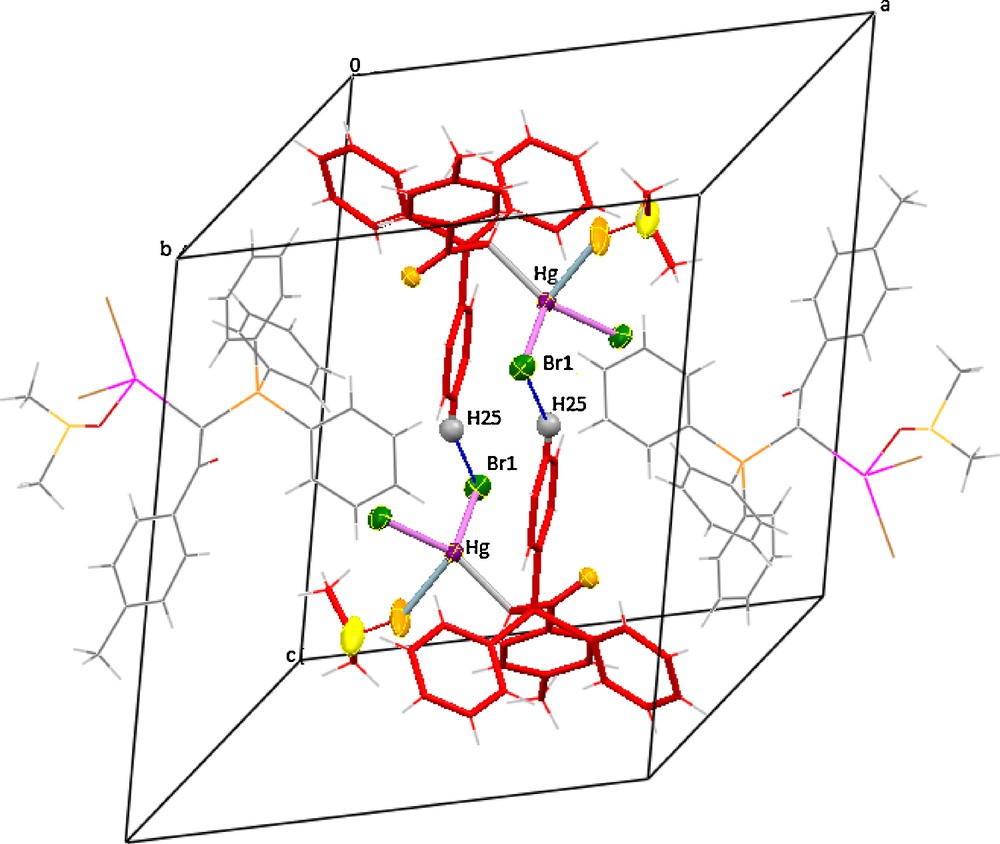
A representation of complex 6 packing showing the association of the adjacent molecules through H (25) Phenyl….Br (1) interactions. Dotted lines represent the short contacts. For visualization of colors, see the online version of this article.
The stabilized resonance structure for the parent ylides is destroyed by the complex formation 6; thus, the C(H)C bond length 1.498(10) Å is significantly longer than the corresponding distance found in the similar uncomplexed phosphorane (1.407(8) Å [28] and 1.401(2) Å) [29]. On the other hand, the bond length of PC(H) in the similar ylide is 1.7194(17) Å [30], which shows that the corresponding bond is considerably elongated to 1.788(7) Å. The C-coordination of the title ylide is in contrast to the O-coordination of the phosphorus ylide Ph3PC(COMe)(COPh) (ABPPY) in a different Hg(II) complex [30]. The difference in coordination mode between ABPPY and these ylides to Hg(II) can be rationalized in terms of the electronic properties and steric requirements of the ylides. The lower electronic density at the ylidic C atom in doubly stabilized ylides compared to simple stabilized ylides has been calculated by DFT (density functional theory) methods recently [31]. It was also demonstrated in the same paper that these factors are not solely responsible for the bonding properties of doubly stabilized ylides. For “simple”stabilized ylides, the C- vs. O-bonding is also a very delicate balance of steric and electronic properties [32]. In this balance it is necessary to consider not only the size and shape of the ligand in the final bonding mode, but also the electronic nature of the metal (Pd, Pt, Ru, Au, etc.) and the donor atoms (C, O, N, etc.) and even the position of the coordination site (trans to a C atom, trans to a N atom, trans to an O atom, and so on). All these facts must be considered as a whole in order to account for the final bonding mode of a given ylide. The nucleophilicity of the carbanion in ABPPY is less than in our ylides; this is due to the additional delocalization of the ylide electron density in ABPPY, which is facilitated by the second carbonyl group. This will reduce the ability of ABPPY to bind via the ylidic carbon. Belluco et al. have studied steric influences on the coordination modes of ylide molecules to Pt(II) systems [8a]. These authors concluded that the preferred coordination mode is via the ylidic carbon, but that steric hindrance around the metal centre or the ylidic carbon will necessitate O-coordination. Indeed, this trend is reflected here, these ylides are slightly less sterically demanding than ABPPY and are C-coordinated to Hg(II).
Literature data show that the coordination mode of dimethylsulfoxide (DMSO) to relatively soft metal atoms depends on both electronic and steric factors deriving from the DMSO moderate π-acceptor properties and its greater steric demand, in the case of S-bonding [33]. In the case of ruthenium(II) complexes, coordination through sulfur (DMSOS) seems to be preferred over coordination through oxygen (DMSOO) to get stable species, unless ligand overcrowding occurs [34] or DMSO is trans to strong p-acceptors like CO [35] and NO [36]. Although on the basis of the Hard-Soft-Acid-Base principle, S-bonding is preferred by ‘soft’ metal ions, but with bulky ligands like present phosphorus ylides, steric effects can induce O-bonding. It may also be seen that for ‘soft’ metal ions, such as Ag(I), Cd(II), and Hg(II), there is evidence of a prevalence of O-bonded species, even in the absence of π-accepting or bulky ligands. This suggests that a particular electronic structure is required in order to favor the metal–sulfur bond over the metal–oxygen one [33]. Bulkiness of present ylides and the absence of the above influences in the structure shown on Fig. 2 lead to preferring O-bonding. An overestimate is found for the calculated S–O distance in ‘free’ DMSO (1.509 Å), which results in a value 0.018 Å longer than the experimental reference one of 1.492(1) Å [37]. The significant elongation of the S–O distance upon O-coordination is further confirmed by the recent X-ray structure determination of disulfoxide and related copper(II) complexes, where the average S–O distances are of 1.487(4) Å (free) and 1.520(3) Å (O-bonded) [38]. It is worth noting that in 6 the SO bond distance of 1.412(8) Å is about 0.080 Å lower than the experimental reference value of 1.492(1) Å for free DMSO ligand [37]. To get an idea about the nature of this distance, a Cambridge Structural Database (CSD) search was carried out with the help of the Vista program (version 2.1) in the November 2010 release of the CSD version 5.32 (ref: The Cambridge Crystallographic Data Center, Cambridge, UK, 2010.). The result of this search on S–O distances for O-coordinated DMSO to any metal shows that only 18 complexes have shorter distance than 1.413 Å.
4 Conclusion
The present study describes the synthesis and characterization of mono- and dinuclear Hg(II) and Cd(II) complexes of (p-methylbenzoyl)methylene triphenylphosphorane as a ligand. On the basis of the physicochemical and spectroscopic data, we propose a monodentate C-coordination to the metals, which is further confirmed by the X-ray crystal structure of 6.
Acknowledgment
This work was supported by Bu-Ali Sina University funds. We thank Mr. Zebarjadian for recording NMR spectra.