

1 Introduction
In a primary description, ylides can be represented as α-zwitterionic species where a positively charged heteroatom X with an octet electron count (such as P, S, N) is covalently bonded to a carbanionic center. In this category, phosphonium ylides Fo possessing an easily pyramidalized carbanion stabilized by an adjacent tetrahedral phosphonium center (Scheme 1), received a long-standing attention not only in the field of organic synthesis, but also of fundamental coordination chemistry. Beyond their ubiquitous role in the Wittig-type olefination of carbonyl compounds, phosphonium ylides are indeed intriguing stable electron-rich C-sp3 carbyl ligands of transition metals [1]. They are today attracting a renewed interest from the standpoints of stereochemistry and catalysis [1]. Depending on the charge delocalization exerted by the substituents, ylides are classified in three families constituted by the non-stabilized, the semi-stabilized and the stabilized versions. Due to their intrinsic stability and their ambidentate character (C- vs O- or Ncoordinating modes), complexes of α-stabilized phosphonium ylide ligands have been naturally more exemplified [1].
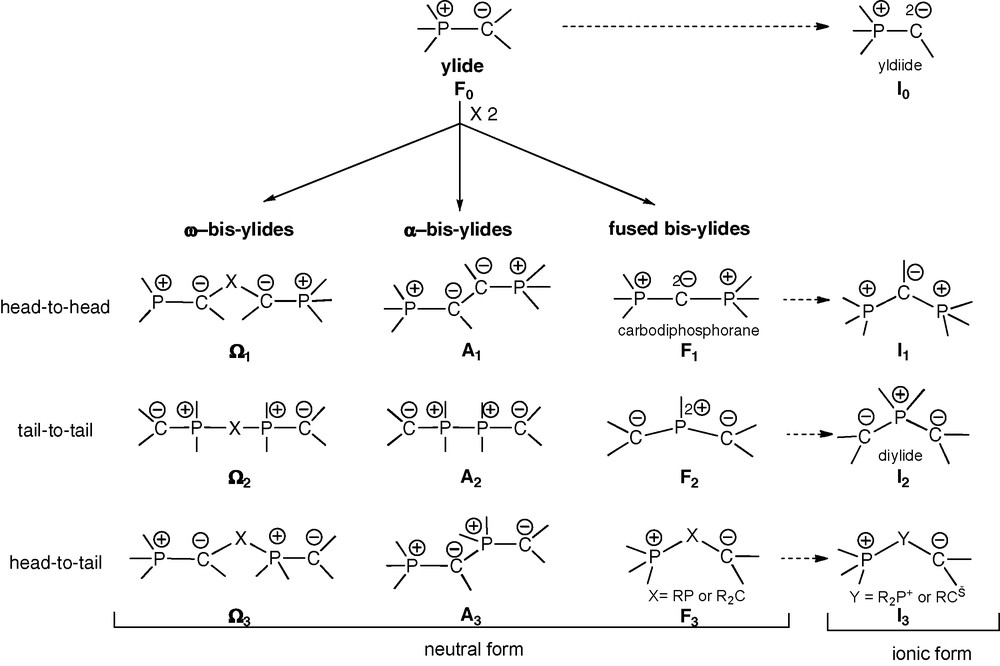
Typology of acyclic and cyclic phosphonium bis-ylides.
Ideally C2-symmetric chelating ligands provide their complexes with particular kinetic stability (entropic factor) and stereochemical control (cis vs trans). Along the same lines as those governing the design of numerous diphosphines, diphosphonium bis-ylides deserve a particular typology. Three types of neutral bis-ylides can thus be distinguished (Scheme 1): the ω-type Ω1- Ω3, the α-type A1–A3 and the fused type F1–F3. The ionic forms I0–I3 of the respective types F0–F3, in particular yldiides I0 and diylides I2, should also be mentioned. Each type gathers three forms depending on the regiochemistry of the coupling between two ylide units F0, consisting each in a carbanionic head and a phosphonium tail: head-to-head, tail-to-tail or head-to-tail.
Among general ω-bis-ylides, the head-to-head form Ω1 is exemplified by β-bis-ylides involving various bridging moieties (X: CO, PR, PR2+, PYZ, P+) [2]. When X = CO or when the ylides moieties are independently stabilized, the ambidentate abilities of such ligands are greatly enhanced. Several ω − bis-ylide ligands of the tail-to-tail form Ω2 (X: (CH2)n, RPPR,…) are also known, including β-representatives (X = RP) [3].
Fused bis-ylides are represented by their head-to-head (C-fused) form F1, corresponding to the carbodiphosphoranes [4]. These species contain two cumulated ylide functions, with a formally divalent central carbon atom with two negative charges stabilized by two phosphonio groups. The presence of two electron pairs at the central carbon atom induces both a bent structure and a very strong nucleophilicity. Carbodiphosphoranes can be regarded as neutral stabilized yldiides of type I0, where a generic C-substituent is replaced by a phosphonium group [5]. Due to their strong nucleophilicity, carbodiphosphoranes are poorly exemplified and undergo easy protonation or alkylation to the corresponding β-diphosphonium monoylides of type I1.
Tail-to-tail (P-fused) bis-ylides of the F2 type, first exemplified by Appel et al. in 1982, correspond to σ3λ5-bis-(methylene)phosphorane derivatives [6]. According to Schoeller's theoretical studies, these polarized π-systems, isoelectronic to allyl anions, possess a weakly pyramidalized phosphorus atom [7]. Their structural and reactivity features are, however, basically similar to those of yldiides I0. Ionic P-alkylated versions of bis-ylides F2 are the so-called diylides I2, acting as stable ligands in transition metal complexes [8] possibly serving as catalyst precursors [8b]. The head-to-tail form F3, with a neutral sp3-C or -P center, is only a formal bis-ylide, i.e., a β-zwitterionic form where the ylidic character is no longer apparent.
In the α-bis-ylides series, the head-to-head (A1) and tail-to-tail (A2) forms exhibit strong C−/C− and P+/P+ electrostatic repulsion, respectively (Scheme 1). Head-to-head representatives A1 may behave as genuine carbene transfer reagents upon coordination: cleavage of a PC bond leads to an ylido-carbene and a phosphino group anchoring to the metal center [9]. The tail-to-tail form A2 was first exemplified by Karsch et al., in 1988 [10]. In these species, both the repulsion of the adjacent cationic phosphorus atoms and the steric bulk of the carbanionic substituents (Me3Si) may explain the observed length of the P+–P+ bond (2.278(1) Å), even longer than the P–P bond between five-coordinate P atoms in a bis-phosphorane (2.264(2) Å). To the best of our knowledge, the sole example of fully characterized head-to-tail α-bis-ylide form A3, with a triple charge alternance (Scheme 1), has been reported by Schmidbaur et al. in 1980 [11].
In contrast to linear diphosphonium bis-ylides (Scheme 1), their cyclic versions have attracted less attention [12]. The case of cyclic head-to-head β-, α- and fused bis-ylide types (B1c, A1c, F1c, respectively) is hereafter systematically addressed (Scheme 2). The corresponding diphosphonium precursors based on the rigid, cis-spacing, insulating 1,2-phenylene bridge have been recently shown to be easily accessible [13].
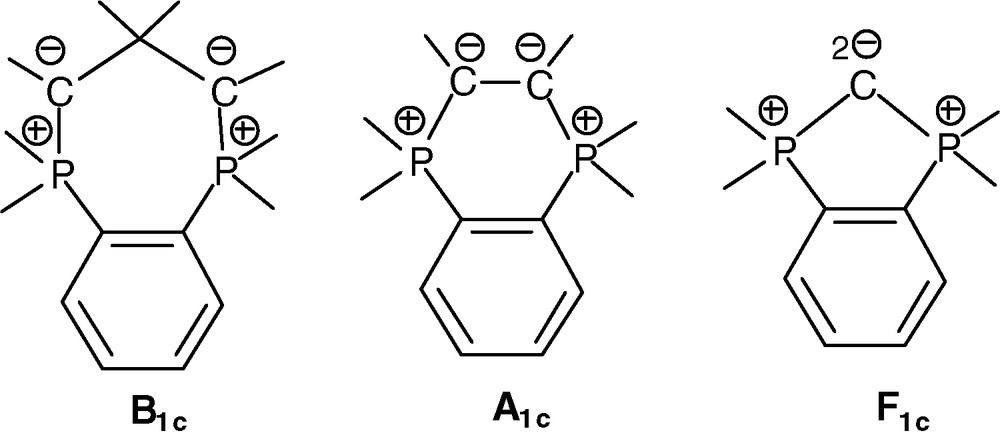
Cyclic head-to-head β-, α- and fused bis-ylides on the o-phenylene framework.
2 Results and discussion
2.1 Non-stabilized head-to-head fused bis-ylides of cyclic diphosphoniums
The diphosphonium 1.Br2 was first obtained by Schmidbaur et al. in 61% yield by refluxing o-bis(diphenylphosphino)benzene (o-dppb) and dibromomethane in toluene over 10 days [14]. We recently reported that replacing CH2Br2 by CH2I2 allowed for the isolation of the analogous diphosphonium 1.I2 in 96% yield over 2 days only (Scheme 3) [13]. A rapid H/D exchange of the methylene protons was observed in the 1H NMR spectrum of 1.I2 in acidic deuterated solvents: the acidity of these protons (δ1H(P+CH2P+) = +5.38 ppm, t, 2JPH = 11.7 Hz) suggests the possible existence of the di-conjugated base, namely the cyclic carbodiphosphorane 2. And indeed, according to Schmidbaur's results, the treatment of 1.Br2 with an ylide base (Et3P=CHMe) in THF at −78 °C led to the formation of the expected cyclic C-fused bis-ylide 2 [14]. This carbodiphosphorane was identified by 31P and 1H NMR spectroscopy at −30 °C, and by the regeneration of the starting diphosphonium 1.Br2 upon quenching with one equivalent of HBr. The thermal unstability of 2 is naturally explained by geometrical and electronic factors; the acute PCP angle in the five membered ring (ca 108°) indeed enforces the sp3 (vs sp) character of the central C atom, thus increasing the weight of the doubly zwitterionic resonance form, and finally a destabilizing charge separation.
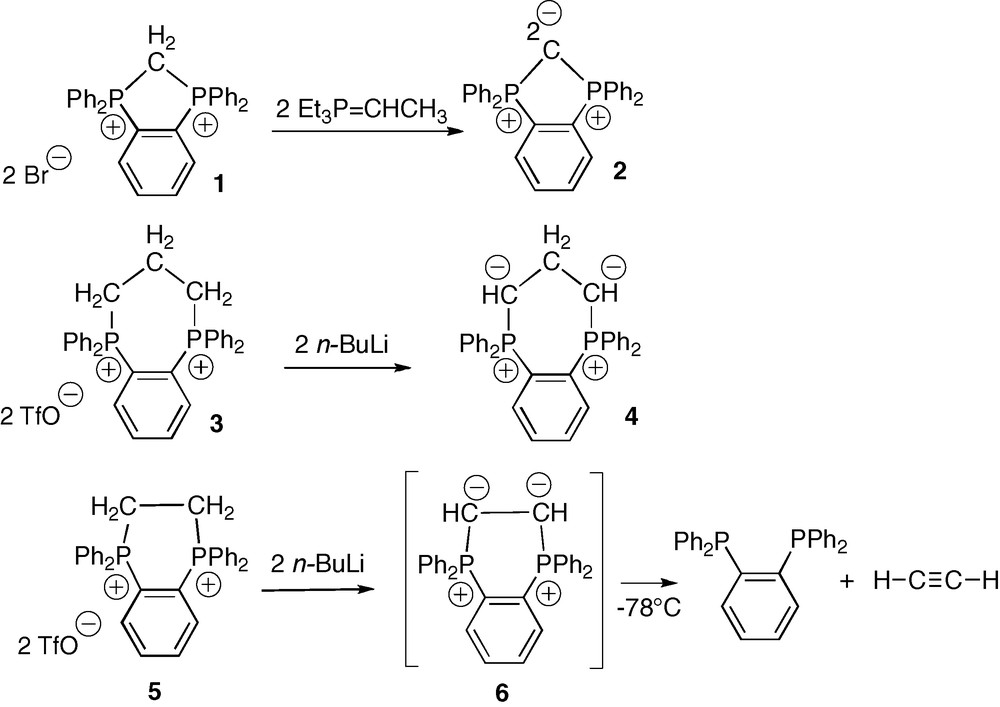
Deprotonation of the cyclic diphosphoniums 1, 3 and 5.
2.2 Non-stabilized head-to-head β-bis-ylides of cyclic diphosphoniums
Elongation of the aliphatic carbon chain between the ylidic carbons and electron delocalization over two carbon atoms instead of one, should result in both geometrical and electronic relaxation. Following this principle, it was recently shown that addition of two equivalents of n-BuLi in THF at low temperature to the less strained diphosphonium 3.(TfO)2 afforded the corresponding cyclic head-to-head β-bis-ylide 4 (Scheme 3) [15]. The structure of 4 was unambiguously established by multi-nuclear NMR spectroscopy, and in particular by the presence of a unique 31P signal at +24.4 ppm, which confirmed the onio character and the equivalence of the two phosphorus atoms. In the 1H and 13C NMR spectra, the simultaneous presence of a triplet of triplets at δH = +0.59 ppm (JHP = 26.8, JHH = 3.3 Hz) and of a triplet at δC = +21.9 ppm (JCP = 4.4 Hz) was assigned to the bridging methylene group of 4. This bis-ylide was reported to act as a strongly donating C,C-chelating ligand of Rh(I) centers [15]. In spite of its strong internal charge repulsion and its low stability (t1/2 ≈ 45 min. at +20 °C), the β-bis-ylide 4 is stable enough to undergo selective transformations. Further bridge shortening from β to α was thus the next challenge.
2.3 Non-stabilized head-to-head α-bis-ylides of cyclic diphosphoniums
The cyclic o-phenylene-diphosphonium 5 with two carbon atoms in the aliphatic bridge, is easily prepared by reaction of o-dppb with 1,2-dichloro-ethane [13]. The corresponding head-to-head α-bis-ylide 6 was targeted by deprotonation of 5 with two equivalents of n-butyllithium in THF at −78 °C. The 31P NMR spectrum indicated the disappearance of 5 (δ31P = +12.6 ppm) and the presence of a much more shielded new signal at δ31P = −10.0 ppm, which was attributed to o-dppb (Scheme 3). Multi-nuclear NMR monitoring in THF-d8 at −78 °C showed the immediate appearance of the o-dppb signal, along with a 1H signal attributable to acetylene, and this even when only a catalytic amount of n-butyllithium was added. Considering the chemical existence of the more strained fused bis-ylide 2 with two geminal negative charges, the non-detection of 6 even at low temperature, can be explained by electrostatic factors, in particular by the enhanced repulsion between the two vicinal negative charges: instantaneous fragmentation results in a neutral o-dppb molecule and a formal α-bis-carbene H–C(:)–C(:)–H in resonance with acetylene (Scheme 4). Phosphonium ylides are indeed recognized as phosphino-carbenoid species [12].

Fragmentation of cyclic head-to-head o-phenylene-diphosphonium α-bis-ylides, R1, R2 = H, CO2Me, CO2Et (see calculated data in Table 1).
This extrusion reaction is supported by the calculation of a strongly exergonic dissociation energy (6 → o-dppb + acetylene: ΔG(298 K) = −21.2 kcal/mol) at the B3PW91/6-31G** level (Scheme 4, Table 1).
Dissociation energies (in kcal/mol) of cyclic head-to-head α-bis-ylides into the corresponding diphosphine (o-dppb) and acetylene derivatives (Scheme 4), calculated at the B3PW91/6-31G** level1.
| R1 = R2 = H | R1 = H, R2 = CO2Me | R1 = R2 = CO2Et | R1 = R2 = CO2Me | R1 = R2 = COPh | |
| ΔH (0 K) | −6.93 | 8.53 | 22.02 | 22.89 | 20.25 |
| ΔHZPE (0 K)a | −9.47 | 6.14 | 19.39 | 20.14 | 17.56 |
| ΔG (298.15 K) | −21.19 | −8.60 | 2.00 | 2.60 | −0.67 |
a Zero-point energy corrected values.
Upon substitution of one of the ylidic carbon atoms of 6 by an ester group (CO2Et or CO2Me), the calculated dissociation energy remains exergonic (ΔG(298 K) = −8.6 kcal/mol), and it becomes slightly endergonic upon substitution of both ylidic carbons (Scheme 4). The stability of such α-bis-ylides, however, cancels if the alkoxycarbonyl groups are replaced by benzoyl groups (Table 1). These trends are in agreement with the relatively “stabilized” character of the corresponding mono-ylides in general. Although the positive Gibbs energies for the dissociation of ester-stabilized α-bis-ylides are quite small in the gas phase, solvent dielectric constant effects are expected to provide additional stabilization of these highly polar species. Experimental efforts in this direction are reported in the next section.
2.4 Stabilized head-to-head α-bis-ylides of cyclic diphosphoniums
Preparation of head-to-head α-stabilized α-bis-ylides is envisaged through the reverse reaction of their fragmentation depicted in Scheme 4. Preparation of the corresponding diphosphonium precursors by adaptation of the previously used method [13] is indeed a priori tricky, and calculations suggest that the transformation is a priori not thermodynamically forbidden (Table 1). Moreover, arylphosphines are known to react with activated unsaturated substrates such as simple acetylenic Michael acceptors [9]. The envisioned process is formally a [2+1+1] cycloaddition which proceed in two steps, initiated by a Michael-type addition [16].
2.4.1 Ester group-stabilized head-to-head α-bis-ylides of cyclic diphosphoniums
As anticipated above, addition of one equivalent of diethyl acetylenedicarboxylate (DEAD) to o-dppb in toluene gave, after 10 min at room temperature, nearly quantitative yield of the targeted bis-ylide 7 as an apparent 70/30 mixture of two stereoisomers (Scheme 5). The occurrence of these two isomers likely results from a restricted rotation about the exocyclic C–CO2Et bonds. Strong delocalization of ylidic electron pairs is indeed classically observed in α-stabilized phosphonium ylides. According to this interpretation, the three Z/Z, E/E and Z/E stereoisomers could, however, be expected.
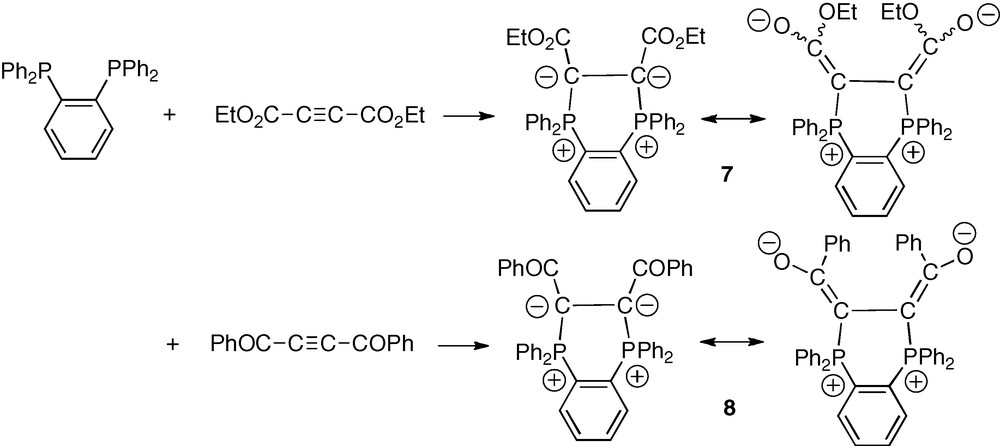
Synthetic approach of α-stabilized diphosphonium α-bis-ylides 7 and 8 from o-dppb.
The structure of 7 in CD3CN solution was first assigned on the basis of the observed 31P NMR chemical shifts of two broad singlets at δP = +0.52 and +2.21 ppm, which are in the typical range for α-stabilized phosphonium ylides. The structure of 7 was then confirmed by 1H and 13C NMR spectroscopy and by mass spectrometry (electron spray [MH]+: 617.2).
A variable temperature 31P NMR experiment indicated a reversible coalescence of the two broad signals at +60 °C corresponding to an activation barrier of ca. 15 kcal/mol, thus demonstrating the dynamic behavior of the different isomers of 7.
By decreasing the temperature to 0 °C, decoalescence in four broad singlets was observed, two of them having the same integral. These signals can thus be globally assigned to the expected three stereoisomers of bis-ylide 7 in a 41/22/37 ratio with respective chemical shifts δPa = +0.36 and δPa’ = +2.40 ppm/δPb = +0.60 ppm/δPc = +2.23 ppm (Fig. 1). Due to dissymmetry, the Z/E isomer exhibits two non-equivalent phosphorus atoms and is therefore assigned to the set of the Pa and Pa’ signals. In contrast, the symmetric Z/Z and E/E isomers exhibit only one type of phosphorus atom and are globally assigned to the Pb and Pc signals. Considering both attractive P+/O− and repulsive O−/O− electrostatic interactions, the E/E isomer is expected to be more stable than the Z/Z isomer: the major signal at δPc = +2.23 ppm (37%) is therefore tentatively assigned to the E/E configuration, while the minor signal at δPb = +0.60 ppm (22%) would correspond to the Z/Z configuration.

Variable temperature 31P NMR spectra of the bis-ylide 7 in CD3CN at 202.53 MHz. For assignment, see text.
The structure of the three stereoisomers of bis-ylide 7 has been investigated at the B3PW91/6-31G** level of calculation. A crude conformational analysis on the CO2Me- substituted bis-ylide model allowed to rule out Cs-symmetric conformers with planar or boat conformations of the aliphatic ring: these conformers are indeed more than 10 kcal/mol higher in energy than C2-symmetric conformers with a twisted aliphatic ring. Conformational analysis of the bis-ylide 7 itself restricted to C2 symmetry yielded several equilibrium structures of quasi-degenerate energies. As anticipated experimentally, the three most stable conformers rank in the energetic order: Z/E < E/E < Z/Z (Table 2). This confirms the assignment empirically proposed on the basis of elementary electrostatic considerations (see above): Z/Z isomer at δPb = +0.60 ppm (22%); E/E isomer at δPc = +2.23 ppm (37%).
Calculated structures of the three stereoisomers of the head-to-head α-bis-ylide 7 (B3PW91/6-31G**).a 31P NMR chemical shifts calculated at the B3PW91/6-31+G** level. For the E/E isomer, the given value is the mean of the calculated chemical shifts (in square brackets) for the frozen optimized C1-symmetric structure.
| Geometry | Z/E | E/E | Z/Z |
| Symmetry | C1 | C1 | C2 |
| Relative energy (kcal/mol) | 0.0 | 0.28 | 0.57 |
| Calcd. 31P δ | −11.9 | −8.57 | −7.54 |
| (ppm)a | 3.9 | [(−16.8 + 0.33)/2] | |
| Exptl. 31P δ (ppm) | 0.36 2.40 | 2.23 | 0.60 |
The 31P NMR chemical shifts of the three stereoisomers of 7 have been calculated at the B3PW91/6-31+G** level (Table 2). The chemical shift values obtained for the inequivalent phosphorus atoms of the E/E isomer in its frozen optimized C1-symmetry have been averaged to regenerate the apparent symmetry due their respective fluxionality at the NMR time scale. The calculated 31P NMR chemical shifts are systematically more shielded than the experimental values. However, although the extreme chemical shifts calculated for the Z/E isomer (−11.9 and 3.9 ppm) correspond to the extreme chemical shifts observed for the same isomer (0.36 and 2.40 ppm), the rigid approximation is not sufficient for reproducing the relative experimental values with further accuracy. This is explained by the narrow range of chemical shifts observed in all the three isomers (Δ = 2.40–0.36 ≈ 2 ppm). Calculations of 31P NMR chemical shifts of strained polar bis-ylides appear therefore less straightforward than those of related diphosphoniums [13].
2.4.2 Keto group-stabilized head-to-head α-bis-ylides of cyclic diphosphoniums
The procedure employed with the DEAD substrate was applied with another electron-poor symmetric alkyne, the 1,4-diphenylbut-2-yne-1,4-dione [17]. Reaction of this diketone with o-dppb in toluene at room temperature over 10 min thus afforded the corresponding cyclic α-bis-ylide 8 in nearly quantitative yield (Scheme 5). The bis-ylide was obtained as a single stereoisomer as indicated by the observation of a unique 31P NMR signal at δP = −0.08 ppm. A variable temperature 31P NMR experiment (−40 → +50 °C) did not indicate any significant change, except a slight sharpening of the signal at low temperatures which can be due to a higher rigidity of the structure of the diketo-bis-ylide 8 as compared to that of the bis(ethoxycarbonyl) bis-ylide 7 (section 2.5). On the basis of empirical geometrical and electrostatic considerations, the configuration of the observed stereoisomer of 8 is assigned to Z/Z, with two cis-Ph2P+–C = C–O− stereogenic units (Scheme 5).
The structure of the three stereoisomers of bis-ylide 8 has been calculated at the B3PW91/6-31G** level. In contrast to the bis-ylide 7, but in agreement with the above-proposed assignment, the Z/Z stereoisomer of C2 symmetry is found much more stable than the Z/E and E/E isomers, lying 4.95 and 10.5 kcal/mol higher in energy, respectively (Fig. 2). The calculated 31P NMR chemical shift of 8 (+6.6 ppm at the B3PW91/6-31+G** level) is quite shielded for a P(IV) nucleus, in qualitative agreement with the experimental value (−0.1 ppm).
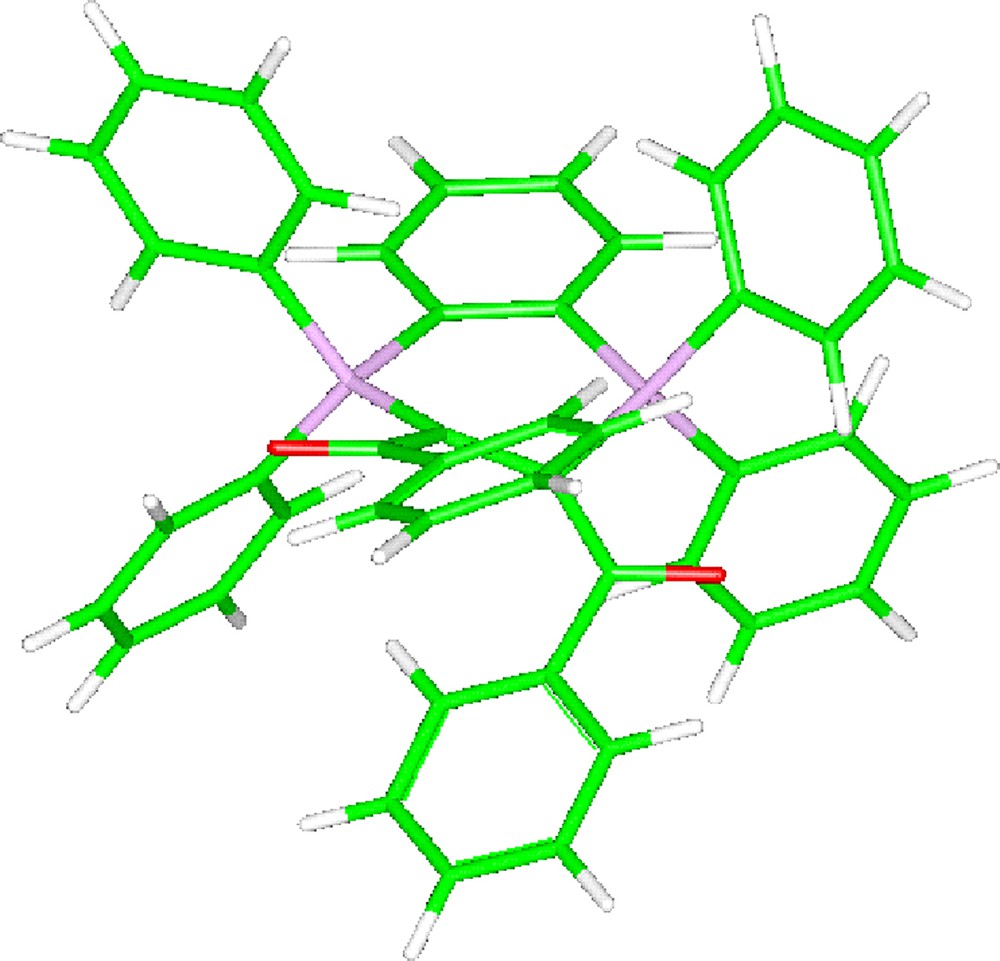
Calculated structure of the Z/Z stereoisomer of the α-bis-ylide 8 (B3PW91/6-31G**).
3 Conclusion
The synthesis and stability of diphosphoniums head-to-head bis-ylides constrained in five-, six- and seven-membered rings have been systematically compared. In the non-stabilized series, it has been shown that while the fused bis-ylide 2 and the β-bisylide 4 are relatively stable, the α-bisylide 6 is unstable due to the repulsion of two vicinal negative charges and undergoes spontaneous fragmentation to o-dppb and acetylene. Introducing electron-withdrawing substituents at the two ylidic carbon atoms allowed for the isolation of stabilized α-bis-ylides 7 and 8. The latter were obtained by a formal [2+1+1] reaction of o-dppb with electron-poor alkynes following a stepwise mechanism. The stabilization effects and the stereochemical behaviour of the α-bis-ylides have been reproduced and analyzed at the theoretical level. In particular, it has been shown that, by contrast to unsubstituted parents, α-bis-ylides stabilized by ester groups exhibit endergonic dissociation. These results illustrate how electrostatic effects (e.g., repulsion between adjacent negative charges) may govern reactivity in geometrically strained frameworks [13].
4 Experimental section
4.1 General remarks
Toluene and diethyl ether were dried and distilled over sodium/benzophenone. All other reagents were used as commercially available. All reactions were carried out under argon atmosphere, using schlenk and vacuum line techniques. The following analytical instruments were used: Bruker ARX 250, DPX 300, AV 500. NMR chemical shifts δ are in ppm, with positive values to high frequency relative to the tetramethylsilane reference for 1H and 13C and to H3PO4 for 31P.
7. To a solution of 1,2-bis(diphenylphosphino)benzene (o-dppb) (0.45 g, 1.0 mmol) in dry toluene (10.0 mL) at room temperature was added diethyl acetylenedicarboxylate (DEAD) (0.33 mL, 2.05 mmol). The reaction mixture was then stirred for 10 min. After evaporation of the solvent under vacuum, the reddish-yellow solid residue was washed by dry diethyl ether (2 × 20 mL) affording 7 as a yellow solid product (yield: 0.60 g, 98%).
31P NMR (CD3CN, 25 °C): δ = +0.52 (s), +2.21 (s) ppm; 31P NMR (CD3CN, 0 °C): δ = +0.36 (s) and +2.40 (s); +0.60 (s), +2.23 (s) ppm. 1H NMR (CD3CN, 0 °C): δ = 0.36 (t, JHH = 7.1 Hz, CH3), 0.41 (broad t, JHH = 7.0 Hz, CH3), 1.07 (broad t, JHH = 7.0 Hz, CH3), 1.11 (broad t, JHH = 6.9 Hz, CH3), 3.33 (m, CH2), 3.54 (m, CH2), 3.78 (m, CH2), 7.18–7.37 (m, Har), 7.44–7.69 (m, Har), 8.12–8.28 (m, Har). 13C NMR (CD3CN, 0 °C): δ = 13.3 (s, CH3), 13.4 (s, CH3), 14.8 (s, CH3), 14.9 (s, CH3), 56.5 (s, CH2), 56.6 (s, CH2), 56.9 (s, CH2), 128.1 (m, CHar, Car), 128.3 (d, JCP = 12.2 Hz, CHar), 128.8 (d, JCP = 15.3 Hz, CHar), 130.2 (d, JCP = 88.1 Hz, Car), 130.9–131.8 (m, CHar, Car), 132.2 (s, CHar), 133.1 (d, JCP = 10.0 Hz, CHar), 133.3 (d, JCP = 10.0 Hz, CHar), 133.7 (m, CHar), 134.4 (pseudo ttt, JCP = 7.6 and 24.0 Hz, CHar), 169.5 (d, JCP = 25.7 Hz, CO), 170.1 (d, JCP = 28.4 Hz, CO), 170.5 (d, JCP = 27.4 Hz, CO), 170.6 (d, JCP = 27.0 Hz, CO). MS (ES+) m/z 617.2 [MH]+. HRMS (ES+) calculated for C38H35O4P2: 617.2011; found 617.2037.
8. To a solution of 1,2-bis(diphenylphosphino)benzene (o-dppbo) (0.45 g, 1.0 mmol) in dry toluene (10.0 mL) at room temperature was added 1,4-diphenylbut-2-yne-1,4-dione (0.48 g, 2.05 mmol). The reaction mixture was then stirred for 10 min. After evaporation of the solvent under vacuum, the reddish-yellow solid residue was washed by dry diethyl ether (2 × 20 ml) giving 8 as a yellow solid product (yield: 0.61 g, 90%).
31P NMR (CD3CN, 25 °C): δ = −0.08 ppm; 31P NMR (CD3CN, −40 °C): δ = +0.24 ppm. 1H NMR (CD3CN, −40 °C): δ = 6.42 (d, JHH = 6.7 Hz, 4H, Har), 6.68 (m, 4H, Har), 7.00 (m, 2H, Har), 7.17–7.29 (m, 4H, Har), 7.40 (m, 3H, Har), 7.50 (m, 1H, Har), 7.58 (m, 2H, Har), 7.66 (m, 1H, Har), 7.77–7.90 (m, 9H, Har), 8.11 (m, 4H, Har). 13C NMR (CD3CN, −40 °C): δ = 124.4 (d, JCP = 73.0 Hz, Car), 126.5 (brs, CHar), 128.1 (brs, CHar), 128.8–129.7 (m, CHar, Car), 130.9 (brs, CHar), 131.7 (brs, CHar), 131.9 (brs, CHar), 132.7 (brs, CHar), 133.5 (m, CHar), 136.1 (brs, CHar), 141.9 (brs, Car), 185.7 (pseudo t, JCP = 9.4 Hz, CO). MS (ES+): m/z 681.2 [MH]+; HRMS (ES+) calculated for C46H35O2P2 681.2112; found, 681.2115.
4.2 Computational details
Geometries were fully optimized at the B3PW91/6-31G** level of calculation using Gaussian 03 [18]. Vibrational analysis was performed at the same level of calculation as the geometry optimization. The magnetic shielding tensor was calculated at the B3PW91/6-31+G** level using the gauge-independent atomic orbital (GIAO) method implemented in Gaussian 03 [18]. The 31P NMR chemical shifts were estimated with respect to the usual H3PO4 reference.
Acknowledgements
The authors thank the French ministère de l’Enseignement supérieur et de la recherche, the French Centre national de la recherche scientifique, the ANR program (ANR-08-JCJC-0137-01) for financial support. The authors also thank Calcul intensif en Midi-Pyrénées (CALMIP, Toulouse, France), institut du développement et des ressources en informatique scientifique (IDRIS, Orsay, France) and centre informatique de l’enseignement supérieur (CINES, Montpellier, France) for computing facilities. M.A. thanks UNESCO, the French Consulate in East Jerusalem, and the conseil régional de Midi-Pyrénées for financial support.
Thanks are also due to one of the referees of this article who draw our attention to the point mentioned in reference [16].
Appendix A Supplementary data
Cartesian coordinates of the calculated structures of the various stereoisomers of 7 (Table 2) and of the Z/Z stereoisomer of 8 (Fig. 2). This material is available free of charge via the Internet at: http://www.sciencedirect.com.