

1 Introduction
Intrinsic conducting polymers with conjugated double bonds are newly considered as a very important class of electroactive and photoactive materials. Some of these polymers exhibit physical properties that lend them to the development of display devices, transistors, sensors, etc. [1–3]. These applications have received a great deal of attention from both academic and industrial research centers [4–7]. During the past decade, thiophene-based electronic materials have been extensively investigated. The ease in chemical modification of the structures of these materials can potentially allow us to fine-tune their optical and electronic properties [8–12]. These properties strongly depend upon the degree of electronic delocalization present in such materials, effective conjugated length (ECL), and introduction of substituents at specific positions. However, a significant drawback of oligothiophenes is their low fluorescence and poor stability with respect to their substituted derivatives [13].
As far as conducting polyheterocyclic compounds are concerned, not only do substituents at the 3-position of the monomer rings prevent the undesirable α–β couplings that decrease the conjugated length and solubility of the polymers, but also the substitution plays an important role in the electrical and electrooptical properties of the polymers [14]. Conducting polymers with modified solubility in some industrially important solvents have recently been synthesized by introducing alkyl groups or other substituents on the monomer rings [15,16]. Since a conducting polymer has a π-conjugate system along the polymer chain, both the electronic and structural properties of a substituent contribute to the delocalization of the polymer π-conjugate system, while its steric factor, which is important for large substituents, is usually not determining [17–21]. In this way, introduction of an electron-donating substituent at the 3-position of the thiophene ring might be an interesting strategy to prevent defects, and withhold or even surpass the desirable properties of polymers.
Theoretical calculations have recently been employed to investigate different aspects related to the molecular and electronic structures of thiophene and its 3-position substituted derivatives using ab initio and DFT calculations at the Hartree-Fock (HF) and B3LYP levels, respectively [22–25]. Theoretical studies for highly conjugated systems have been extensively used by the DFT calculations. For this purpose, calculations were performed on the oligomers containing some monomer units in both the neutral and doped states [26–29]. In our previous works [30,31], we studied a series of haloalkylpyrroles as potential monomers for the synthesis of conductive polymers, and the corresponding oligomers with modified physical and electrical characteristics with respect to the oligomers consisted of pyrrole monomers as reference compounds for all comparative studies. These studies confirmed that the choice of a proper substitution on the heterocyclic monomers can effectively change the mechanical properties and then electrical conductivity of the corresponding polymers.
Poly(3-alkylthiophene) compounds represent a class of conducting polymers that are soluble and processable, and yet retain the magnitude of electrical conductivity of the insoluble parent, polythiophene. In this work, we studied the structural and electronic properties of 3-alkylthiophenes containing oligomers, as obtained with the aid of DFT calculations, and compared this data with the corresponding properties of electron-donating substituents such as the methyl, ethyl, and n-propyl groups at the 3-position of the thiophene ring. To this end, we investigated characteristics of the π-conjugate system and bond length alternation pattern, charge and spin density distributions, HOMO-LUMO gaps (HLGs), and ionization potential (IP) energies of a series of OATs including mono-, di-, tri-, tetra-, penta-, and hexamers.
2 Computational details
The ground-state geometry of each oligomer studied in this work was fully optimized using gradient procedures at the Hartree–Fock and hybrid density functional B3LYP levels of theory. Our previous studies [30,31] showed that 6-31G** is the best basis set considering computational times and our available hardware facilities for highly conjugated systems. Calculations on the electronic ground state were carried out using the DFT and HF methods, and the values for the difference between the calculated DFT and HF electronic energies, obtained by ΔE = EDFT – EHF, are presented in Table 1. According to this table, the minimization results show that the DFT calculations are more stable than the HF calculations for all the molecules studied. This is due to the fact that electron correlation effects are not included in the HF method. Based on these preliminary studies, DFT-B3LYP level of theory was selected for the present study of OATs using the 6-31G** basis set as implemented in the Gaussian 03 program package [32]. The optimized geometries were calculated for both the neutral and doped states of the oligomers. There was no symmetric constraint on the geometric optimization. The nature of the optimized stationary point was characterized by frequency calculations at the same level of theory with the same basis set. Analysis of the obtained results showed that all the optimized structures were obtained as minima on the potential energy surface without any imaginary frequency. A preliminary basis set test carried out at the RHF level of theory showed that 6-31G** was the best basis set that can be used within our available hardware/software facilities within a reasonable time. The energies of the different states in the relevant geometries were obtained for calculating the IP values using the Koopman theorem [33].
ΔE values (in eV), difference between DFT and HF electronic ground state energies, for all studied molecules.
| n | R = H | R = CH3 | R = C2H5 | R = C3H7 |
| 1 | –1.7115 | –1.9912 | –2.2700 | –2.5488 |
| 2 | –3.3855 | –3.9443 | –4.5017 | –5.0609 |
| 3 | –5.0601 | –6.5451 | –6.9094 | –7.5718 |
| 4 | –6.7348 | –7.8531 | –8.9662 | –10.2015 |
| 5 | –10.0845 | –9.8069 | –11.1992 | –12.7742 |
| 6 | –11.0957 | –11.7612 | –12.4485 | –14.2820 |
3 Results and discussion
3.1 Molecular geometry
The sketch map of the structures for some OATs is depicted in Fig. 1. It is noticeable that the carbon atoms adjacent to the heteroatom in the thiophene ring, α and α′ positions, are branching centers in the electropolymerization process, and thus control the stereochemistry of the polymer chains. Also both the β and β′ carbon atoms are appropriate positions for substitution. However, in this study, the alkyl substitution was fixed on the β-position in all compounds. For easier comparative studies, and similar to the monomer structures, in all oligomer chains, the α-carbon atom is adjacent to the substituent position (β), and the α′-carbon atom is away from it at the head and tail of an oligomer chain. It is known that the well-defined polymer structure contains almost exclusively head-to-tail (HT) conformation, due to the α–α′ couplings between adjacent heterocyclic monomers in the backbone of polymer chains [30,31]. Thus geometry optimization of OATs was performed on the HT conformation at the B3LYP/6-31G** level of theory.
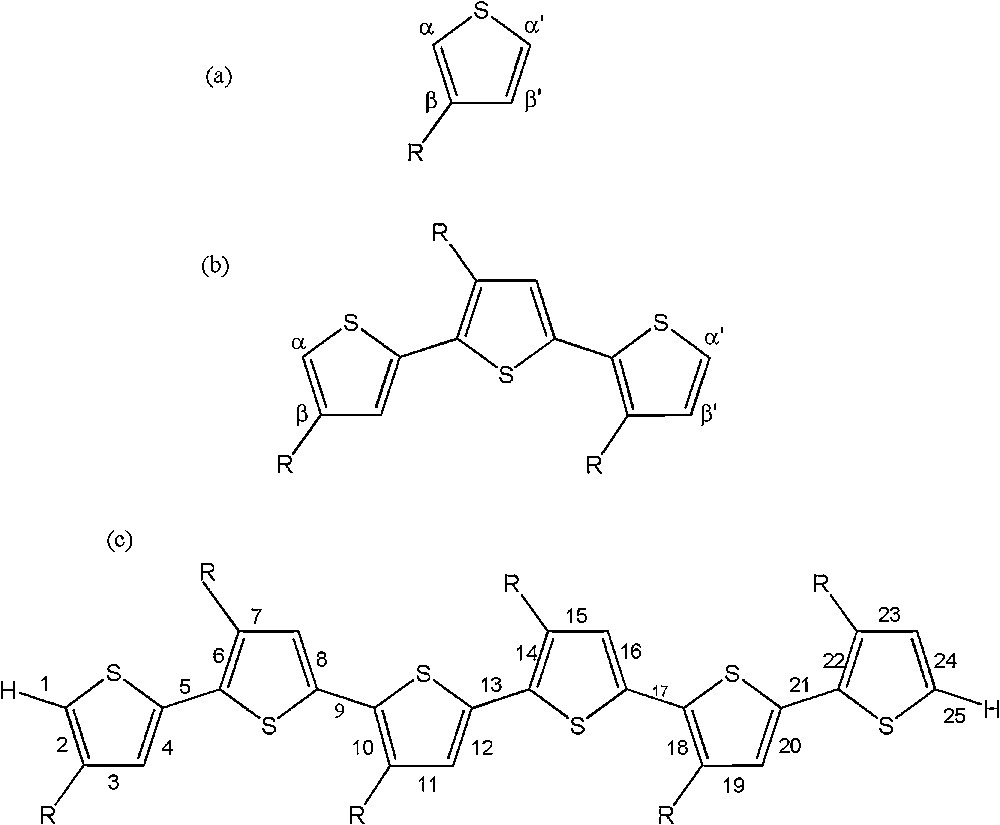
Structure and numbering scheme used for the AT: (a) monomer; (b) trimer; and (c) hexamer, where R stands for CH3, C2H5, and C3H7.
The C–C bond lengths in the conjugated π-system of the optimized oligomers were compared in Fig. 2 for the hexamer type (n = 6), as an example. The alternative apparition of single and double C–C bonds is consistent with an aromatic-like electronic structure for the three substituents, even though the differences found among them indicate that in some cases contribution of the quinoid form to the electronic structure is considerable. The quinoid term refers to a structure in which the inter-ring bond has a greater double bond character than that in the standard aromatic configuration, and it is determined by the quinoid coefficient, fn [34–37]. This geometry parameter is defined as:
| (1) |
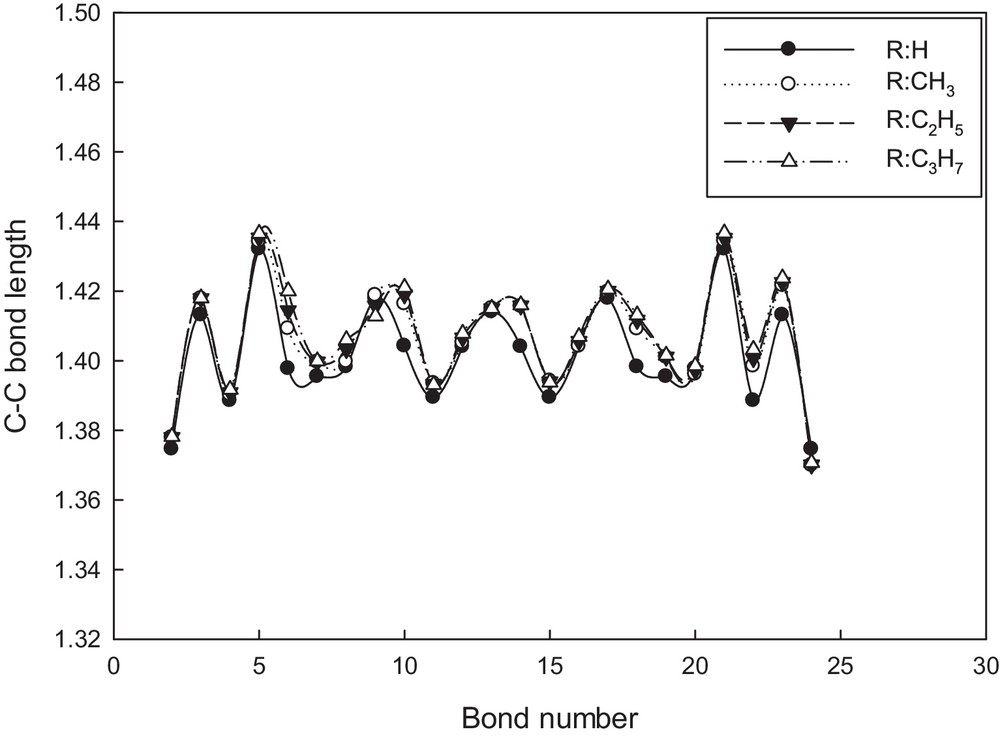
Behavior of C–C bond lengths along the conjugated π-system for hexa(ATs). The data were taken from the molecular structures optimized at B3LYP/6.31G** (Fig. 1c).
Quinoid coefficient value, fn, for C–C bond lengths along the conjugated π-system for OATs.
| Oligomer | R = CH3 | R = C2H5 | R = C3H7 |
| Dimer | 1.0139 | 1.0100 | 1.0135 |
| Trimer | 1.0124 | 1.0107 | 1.0108 |
| Tetramer | 1.0095 | 1.0094 | 1.0093 |
| Pentamer | 1.0073 | 1.0074 | 1.0086 |
| Hexamer | 1.0042 | 1.0049 | 1.0055 |
Dihedral angles defining the torsion between two adjacent building units along the OATs in doped states were collected in Table 3. As shown in this table, all the OATs have a planar configuration. Also the type of alkyl substituent does not have a significant effect on the planarity of oligomers. However, the exact torsion angle value for the methyl substituent is zero. With increase in the repeating units, there is a significant reduction in the inter-ring torsion angle at the center of the oligomer, compared to the outside. It is interesting that the doped oligomers have less torsion angles than the corresponding neutral ones. It is known that the planarity of the polymer chains has an important role in their electric conductivity. In general, crystalline oligothiophenes have been found to be nearly planar as a result of more favorable crystal packing [38]. The DFT-optimized geometries are in agreement with the solid-state structure.
Dihedral angels (in degrees) for the building units along the conjugated π-system of OATs in the doped state.
| Oligomer | R = H | R = CH3 | R = C2H5 | R = C3H7 |
| Dimer | 0.00 | 0.00 | 0.18 | 0.60 |
| Trimer | 0.00 | 0.01 | 2.50 | 3.50 |
| Tetramer | 0.00 | 0.00 | 3.65 | 5.15 |
| Pentamer | 0.00 | 0.01 | 0.85 | 0.60 |
| Hexamer | 0.00 | 0.01 | 0.35 | 1.60 |
3.2 Electronic properties
Since charge density of the α and α′ carbon atoms at both sides of the oligomer chains, as the propagation centers in the growth polymer chains, is an important factor in the polymerization mechanism of a conductive polymer, we investigated qualitatively the “push-pull” effect of substituents on the AT oligomers by the Mulliken population analysis using the net atomic electric charge distribution and spin density. The results obtained for the doped states were summarized in Tables 4 and 5. These data show that both the α and α′ carbon atoms in thiophene and its oligomers have equivalent charge and spin densities, but this equality is altered with the addition of an alkyl substituent to the thiophene ring. For the long oligomer chains, the negative charge on the α-carbon atom, which is close to the substitution position, is higher than that on the α′-carbon atom, which is far from it (Fig. 1). Similarly, spin density is distributed mainly on the α′-carbon atom. This is due to the fact that the negative charge and spin densities are carried by electrons whose charge is negative. The high spin density at the α and α′ positions of the thiophene monomer indicates that these positions are the reactive sites in the monomer, and this also suggests that the monomer linkages in the polymer occur at these positions.
Distribution of charge density on the branching center, α (α′), of OATs in the doped state.
| n | R = H | R = CH3 | R = C2H5 | R = C3H7 |
| 1 | –0.20 (–0.20) | –0.24 (–0.24) | –0.22 (–0.24) | –0.20 (–0.20) |
| 2 | –0.24 (–0.24) | –0.25 (–0.25) | –0.28 (–0.25) | –0.27 (–0.25) |
| 3 | –0.26 (–0.26) | –0.26 (–0.26) | –0.30 (–0.26) | –0.30 (–0.26) |
| 4 | –0.27 (–0.27) | –0.27 (–0.27) | –0.32 (–0.27) | –0.32 (–0.27) |
| 5 | –0.27 (–0.27) | –0.28 (–0.28) | –0.33 (–0.28) | –0.33 (–0.28) |
| 6 | –0.28 (–0.28) | –0.28 (–0.28) | –0.34 (–0.28) | –0.33 (–0.29) |
Distribution of spin density on the branching center, α (α′), of OATs in the doped state.
| n | R = H | R = CH3 | R = C2H5 | R = C3H7 |
| 1 | 0.54 (0.54) | 0.47 (0.45) | 0.48 (0.44) | 0.54 (0.54) |
| 2 | 0.31 (0.31) | 0.36 (0.28) | 0.36 (0.27) | 0.36 (0.27) |
| 3 | 0.21 (0.21) | 0.25 (0.18) | 0.25 (0.17) | 0.25 (0.17) |
| 4 | 0.15 (0.15) | 0.18 (0.12) | 0.18 (0.12) | 0.18 (0.12) |
| 5 | 0.11 (0.11) | 0.14 (0.09) | 0.13 (0.10) | 0.14 (0.09) |
| 6 | 0.08 (0.08) | 0.11 (0.06) | 0.11 (0.07) | 0.10 (0.08) |
Delocalization of the unpaired spin onto the oligomer chain leads to satisfactory resonance structures and improved stabilization of the radical cations. The spin density at the reactive sites (α and α′ positions) decreases when the oligomer chain length increases. Compared to the thiophene monomer, presenting an electron-donating group such as an alkyl group does not alter the spin density of OATs significantly, and therefore, could be expected to allow electropolymerization.
It is useful to examine the highest occupied orbitals and the lowest virtual orbitals for these oligomers and polymers because the relative ordering of the occupied and virtual orbitals provides a reasonable qualitative indication of the excitation properties and of the ability of electron or hole transport [39]. Because the first dipole-allowed electron transitions as well as the strongest electron transitions with largest oscillator strength correspond almost exclusively to the promotion of an electron from HOMO to LUMO, we calculated the HLGs for AT oligomers in both the neutral and doped states at the B3LYP/6-31G** level of theory. The HLG values for the neutral form are shown in Fig. 3. As we can see in this figure, reduction of HLGs for all OATs becomes more obvious with increase in the conjugation length. Also, for brevity, only HLG values for a series of oligo(propylthiophenes) in both states were compared in Fig. 4. As shown in this figure, the HLG values for the doped states are less than those for the neutral ones. Therefore, we may predict the polymers in the doped states with long length of chains to have the highest conductivity.
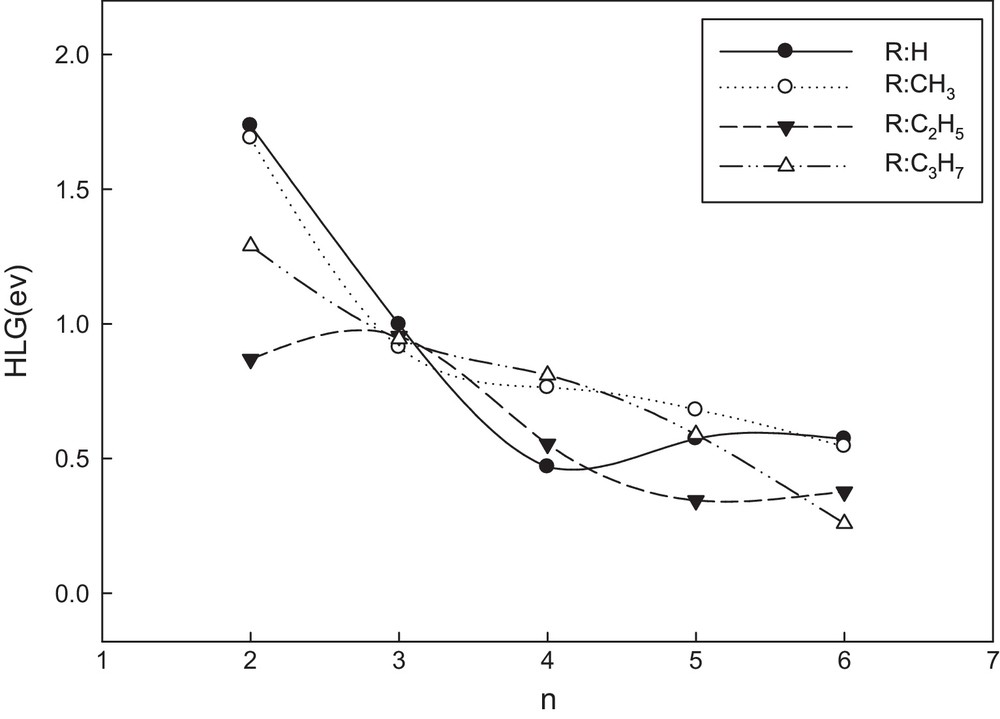
Variation in HLG with the number of units (n) for all alkylthiophene oligomers in the neutral state.

Comparison between HLG values for oligo(propylthiophene) in the neutral and doped states.
As mentioned in the introduction section, efficient injection and transport of both holes and electrons are important parameters for the rational design of optimized light-emitting diodes. The ionization potential values can be used to estimate the energy barrier for the injection of holes into the polymer. Table 6 contains IPs for all the studied molecules. Fig. 5 displays plots of IP values as a function of reciprocal chain lengths for OATs. As far as the whole trend is concerned, with increase in the number of monomers in the oligomer chains (n→∞), the tendency for linearity of the curves increases. We considered all points to plot the regression curves and extrapolate the resulting curves to infinite chain length (n→∞). It was found that as the fraction of AT increased, the IP values decreased, and this corresponds to the improvement in the hole injection properties, and this is also consistent with the trend of changes in the energies of HOMOs. As in the case of HLGs, with increase in the conjugated length, the influence of the substituent type is not considerable.
Ionization potentials values for OATs (in kcal/mol).
| n | R = CH3 | R = C2H5 | R = C3H7 |
| 1 | 6.96 | 6.89 | 6.84 |
| 1 | 5.80 | 5.78 | 5.74 |
| 3 | 5.26 | 5.22 | 5.22 |
| 4 | 4.96 | 4.96 | 4.96 |
| 5 | 4.755 | 4.77 | 4.74 |
| 6 | 4.61 | 4.56 | 4.52 |
| ∞ | 3.804 | 3.75 | 3.74 |
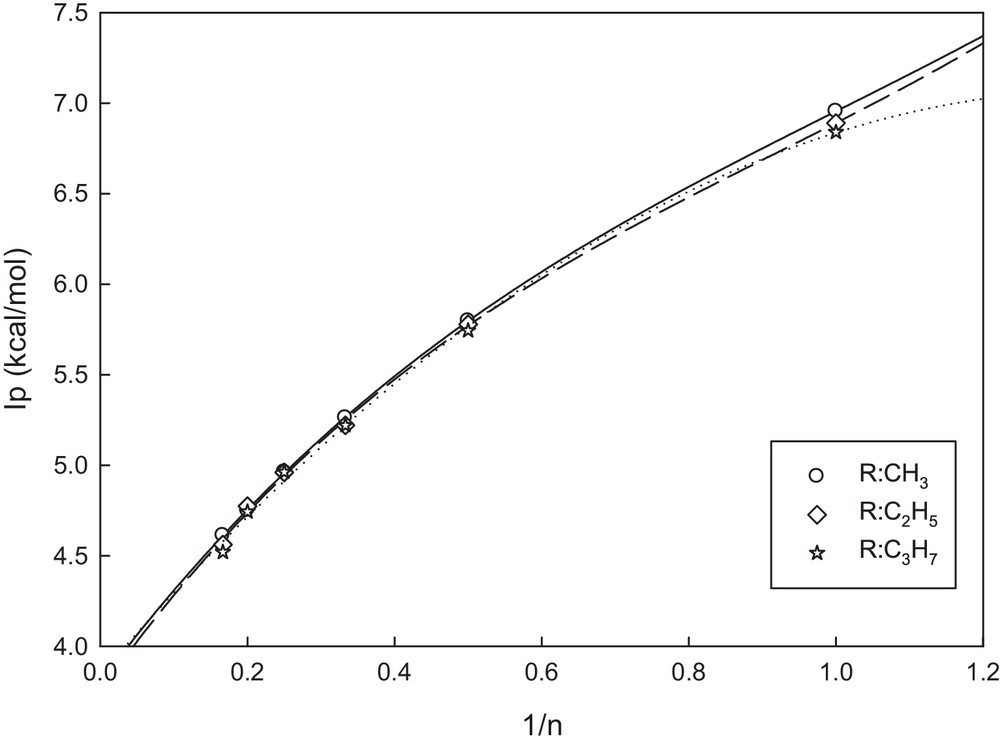
Variation in the IP values as a function of reciprocal chain length with the number of units (n) for AT oligomers in the neutral state.
Our calculations also show that the IP values for these compounds decrease when an electron-donor substituent is introduced onto the thiophene derivatives. It is known that the formation of the intermediate radical cations from the monomers, as the first step in the polymerization mechanism of conductive polymers, has an important role in their polymerization process. This reaction can be considered as an ionization reaction [40–42]. In general, it may be concluded that the initiation step of electropolymerization of AT monomers requires a lower applied potential. In other words, this potential is reduced when the number of repeating units in the oligomer increases. This behavior is in agreement with the lower oxidation potentials measured experimentally [39]. All in all, we may predict that the transporting properties in the polymers are better than those in the oligomers.
4 Conclusion
In this work, the influence of substitution of alkyl groups including methyl, ethyl, and n-propyl groups on the structural and electronic properties of oligomers of alkylthiophenes was investigated using the DFT. Presence of an alkyl substituent on the β-position of the heterocyclic ring alters the charge equilibrium and spin density in the α and α′ positions of the oligomer chains. For the long oligomer chains, the spin and charge density values on the α-carbon atom, which is closer to the substitution position, are higher than those on the α′-carbon atom, which is far from it. The calculations carried out on both the neutral and doped oligomers show that the doped oligomers have more satisfactory structural and electronic characteristics for conducting polymers. The conjugated π-system in the doped oligomers is expanded more, with a higher planarity and the quinoid coefficient close to unit. The obtained results reveal that with increase in the oligomer chain length, the ratio of bond lengths in a conjugated system tends towards unit, corresponding to higher aromaticity. Also the calculated energy gap values between the frontal orbitals and ionization potentials indicate that with increase in the oligomer chain length, the conductivity band decreases. Furthermore, we may predict that the type of alkyl substituent does not have a considerable effect on the electronic properties of the polymers.
Acknowledgement
The authors wish to thank the Shahrood University of Technology for the financial support of this work.