

1 Introduction
Thiacalixarenes are molecules in the calixarene family in which the methylene bridge of the classical calixarenes is replaced with epithio groups [1]. Their ease of preparation and high potential for numerous applications has increased the interest in this class of macrocycles and, in particular, for the preparation of polynuclear metal complexes, an endeavor in which our research group has been involved for several years [2,3]. In a previous publication, we reported the first quantitative syntheses of several mono-O-alkylated-thiacalix[4]arenes in order to break the symmetry of the phenolic crown and thus, obtain a multichelating asymmetric ligand [4]. To increase the complexing power of this mono-O-alkylated ligand, we decided to introduce additional complexing functionality by adding a diaminoethane group via aminolysis of the 28-O-(ethoxycarbonyl)methoxy-2,8,14,20-tetrathiacalix[4]arene 1. The expected products could be used as ligands, but also as intermediates, which could lead to the bridging of two macrocycles.
In this paper, we report the aminolysis of 1 by various diaminoalkanes. Unexpected properties of this family of compounds are presented, such as the formation of colloidal suspensions or self-assembled wires. Finally, the monoamido p-tert-butylthiacalix[4]arene derivatives are used as the precursors to synthesize a novel series of mono-O-bridged bisthiacalix[4]arenes.
2 Results and discussion
The reaction started from 5,11,17,23-tetra-tert-butyl-28-(ethoxycarbonyl)methoxy-2,8,14,20-tetrathiacalix[4]arene 1, which was transformed into 5,11,17,23-tetra-tert-butyl-25-O-[((N-aminoalkyl)aminocarbonyl)-methoxy]thiacalix[4]arene by aminolysis with an excess of an alkyldiamine under atmospheric conditions (Scheme 1).
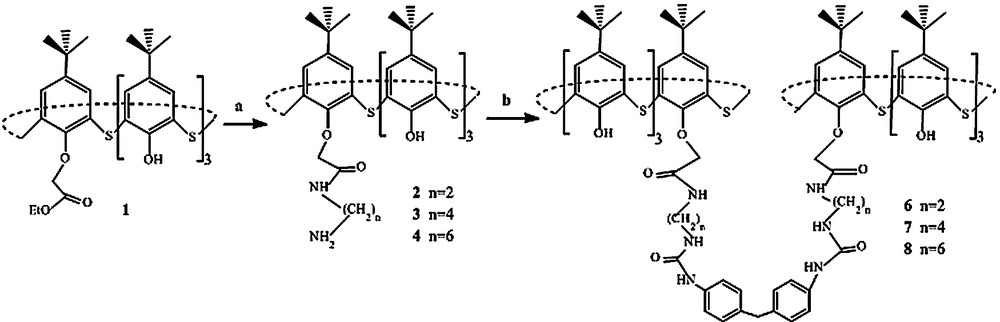
(a) Aminolysis reactions of 5,11,17,23-tetra-tert-butyl-25-O-(ethoxy)methoxy-2,8,14,20-tetrathiacalix[4]arene 1. (b) 4,4′-methylenebis(phenylisocyanate), catalyst (dibutyltin dilaurate), THF, RT, N2.
The 1H NMR spectra of compounds 2–4 reflected the ABBB ring symmetry from the signals at 1.04 ppm due to the tert-butyl groups (2:1:1 ratio). The three signals from the aromatic groups were also indicative of the monosubstituted structure. This prediction was also confirmed by mass spectrometry (MS–ESI). An important feature of this NMR analysis is that signals corresponding to the 4-aminoalkyl groups are absent from the spectrum when CDCl3 is used as the solvent. This could be related to molecules 2–3 self-assembling into meso-structures, like micelles or vesicles (during the precipitation step of the purification, the product is usually found in the form of a colloidal suspension or of a gel). These hypotheses can be validated by measuring the diffusion coefficient from self-assembled structures using diffusion ordered spectroscopy (DOSY). The various textures of the solid obtained support the assumption of the macromolecules undergoing self-assembly on the media. A preliminary characterization carried out by scanning electron microscopy (SEM) revealed an unexpected organization of 2. Indeed, during the purification process, the addition of water to a solution of 2 dissolved in acetone first induces a colloid suspension, then, a gel texture. The self-assembled product is composed of particles having a diameter from 0.01 to 0.5 μm and of a tangle of wires of diameter ranging from 0.1 to 0.4 μm (Fig. 1).

SEM image of structural characterization of 2.
Compared with 3 and 4, compound 2 reacts with an amine, such as triethylamine, for example, in the solid state or in solution by heating, to give compound 5 (Scheme 2).

Cyclization of 2. (i) Heating with triethylamine.
Generally, 2-imidazolines are easily prepared in good yields by the reaction between aldehydes and ethylenediamino molecules or by the cyclization reaction of the acylethylenediamine group, but, to the best of our knowledge, no thermal cyclization reaction with the carbonyl group of the amide function is known for the calixarene family [5]. Compound 5 was synthesized using two different routes: a solid state route and a solution one. Solid state reactions can be used as an efficient method for reducing reaction times. The approach consists in heating the solid state of a macrocycle whose skeleton is thermally stable, whereas its pendant arms are chemically reactive, as we have already demonstrated during the synthesis of thiacalixthiantrene [6]. The structure of compound 5 was solved by single-crystal X-ray diffraction. Suitable crystals for diffraction were obtained by slow diffusion of ethyl acetate into a dichloromethane solution of compound 5, which crystallized in the non-centrosymmetric space group P21212. The asymmetric unit consists of two molecules 5A and 5B, differing in the position of the 2-imidazoline group around the methoxy bond, as shown in Fig. 2. The conformation of each macrocycle is a distorted pinched cone with two opposite aromatics rings tilted towards each other. Another interesting feature of the derivative 5 is represented by its molecular packing. Fig. 3a shows that along the a-axis, the macrocycles pack as “5A–5A” and “5B–5B”, head-to-head to bilayers, the polar O substituents forming the interior of the bilayers. The presence of the tert-butyl head groups precludes the insertion of the short alkyl chains into the macrocyclic cavity. The integrity of the structure is partly assured by multiple hydrogen bonds between 5A–5A and 5B–5B (Fig. 3b).
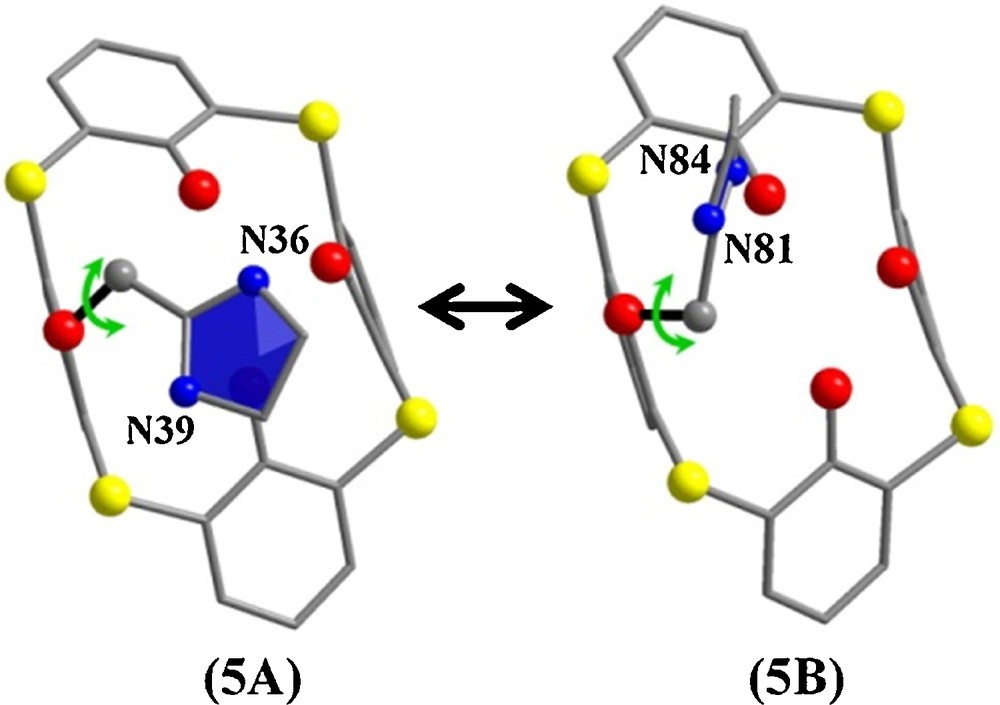
Representation of the two macrocycles 5A and 5B that make up the asymmetric unit (left) and a view showing the intramolecular hydrogen bonds (right). p-tert-Butyl groups and hydrogen atoms are omitted for clarity. Each atom is depicted as follows: S, yellow; O, red; N, blue; C, gray. (For interpretation of references to color, see the online version of this article).
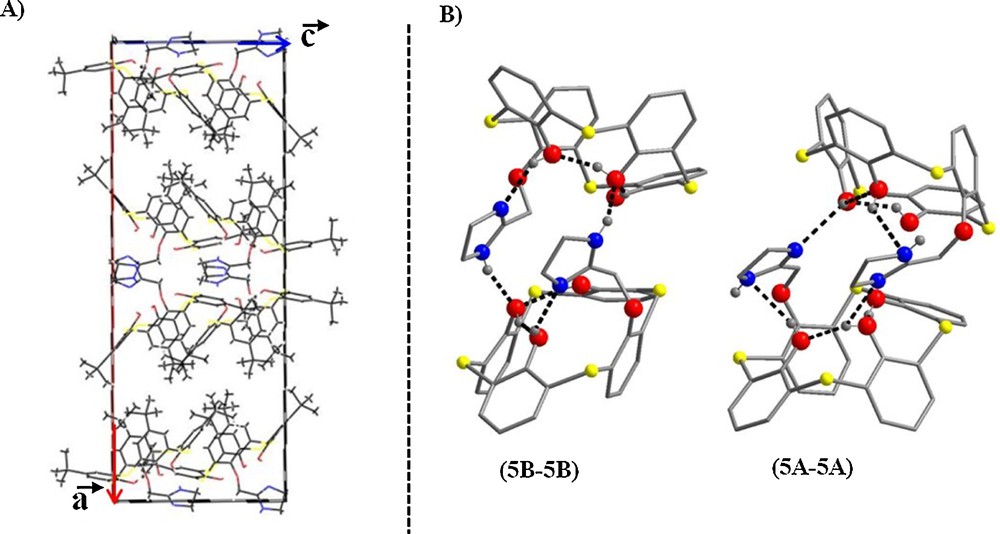
(A) Crystal packing of complex 5, viewed along the b-axis; (B) view showing the intramolecular hydrogen bonds between 5A–5A and 5B–5B depicted in dotted black lines. p-tert-Butyl groups and hydrogen atoms are omitted for clarity. Each atom is depicted as follows: S, yellow; O, red; N, blue; C, gray. (For interpretation of references to color, see the online version of this article).
The choice of the imidazoline group as a linker is not innocent, as it has been shown to be a good ligand for coordination chemistry, in particular, its ability to mimic peptide bonds [7]. The new reaction described herein allows us to consider the design of new types of di-symmetrical receptors in a more versatile manner than is commonly encountered, and that generally possess a higher symmetry in the arrangement of ligands bound around the hydrophilic crown of the macrocycles.
Finally, a series of mono-bridged bisthiacalix[4]arenes has been achieved through the formation of a biuret bond between methylenediphenyl-4,4′-diisocyanate (MDI) and thiacalixarenes 2–4 (Scheme 1). Other ways of connection via peptide bonds have been investigated, without successful results. The synthetic route used is presented in Scheme 1. The synthesis of bisthiacalix[4]arenes 6–8 was confirmed by spectroscopic and analytical data. These compounds have a wide range of functions, conformational flexibility and chemical behaviour, which potentially increase their importance in supramolecular chemistry [8].
3 Conclusion
In conclusion, we have shown that 5,11,17,23-tetra-tert-butyl-25- (ethoxycarbonyl)methoxy-2,8,14,20-tetrathiacalix[4]arene 1 can be used as a starting point in the synthesis of multiple thiacalix[4]arene derivatives. Aminolysis of 1 with various diaminoalkanes lead to the new compounds 2–5. An unexpected self-assembly property of 2 provides promising new perspectives that are currently under study.
4 Experimental
4.1 General remarks
All the chemicals and solvents were used as received (solvents: Carlo Erba RPE; chemicals: Aldrich); all preparations and manipulations were performed under aerobic conditions. The compounds p-tert-butylthiacalix[4]arene (ThiaS) and 5,11,17,23-tetra-tert-butyl-25-(ethoxycarbonyl)methoxy-2,8,14,20-tetrathiacalix[4]arene were synthesized according to the published procedures [1,4]. FT–IR spectra were recorded on a Nicolet 380 spectrometer. Mass spectra were performed on a MicroTOF-QII Bruker mass spectrometer (ESI Source). 1H NMR spectra were run on a Bruker DRX-300 (300 MHz) AVANCE instrument.
4.2 Crystallographic data
A suitable crystal was selected and mounted on a Gemini kappa-geometry diffractometer (Agilent Technologies Ltd, UK) equipped with an Atlas CCD detector and using Mo radiation (λ = 0.71073 Å). Intensities were collected at 100 K by means of the CrysalisPro software [9]. Reflection indexing, unit-cell parameters refinement, Lorentz-polarization correction, peak integration and background determination were carried out with the CrysalisPro software. An analytical absorption correction was applied using the modeled faces of the crystal [10]. The structures were solved by direct methods with SIR97 and the least-square refinement on F2 was achieved with the CRYSTALS software [11]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were all located in a difference map, but those attached to carbon atoms were repositioned geometrically. The H atoms were initially refined with soft restraints on the bond lengths and angles to regularize their geometry (C–H in the range 0.93–0.98 Å, N–H in the range 0.86–0.89 Å and O–H = 0.82 Å and Uiso(H) in the range 1.2–1.5 times Ueq of the parent atom), after which the positions were refined with riding constraints.
4.3 Compound 5
C44H53N2O4S4, fw = 802.16, orthorhombic, P21212, a = 37.411(2) Å, b = 16.378(1) Å, c = 14.067(1) Å, V = 8623.8(9) Å3, Z = 8, 14 933 independent reflections, 13 577 reflections were observed (I > 2.0 σ(I)), R1 = 0.069, wR2 = 0.133 (observed). Crystallographic data reported in this article have been deposited within the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC 881980.
4.4 Typical procedure
4.4.1 Synthesis of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(2-aminoethyl)aminocarbonyl)-methoxy]thiacalix[4]arene 2
5,11,17,23-tetra-tert-Butyl-25-O-[(ethoxycarbonyl)methoxy]-2,8,14,20-tetrathiacalix[4]arene 1 (1 g, 1.24 mmol) was dissolved in 5 mL of 1,2-diaminoethane. The yellow solution was stirred for three days at ambient temperature and 100 mL of a brine solution were added. The resulting precipitate was filtered off and taken up in CHCl3. The organic phase was extracted and washed with H2O (80 mL). The organic layer was dried over MgSO4, and the solvent was evaporated. The product was obtained by precipitation from methanol as a yellowish powder (50–60%) with mp 232 °C. 1H NMR (300 MHz, DMSO): 7.53 (d, 4H, Ar), 7.32 (s, 2H, Ar), 6.33 (s, 2H, Ar), 5.50 (s, 2H,–O–CH2), 3.52 (t, 2H, J = 6.3 Hz,–CH2–NH), 3.02 (t, 2H, J = 6 Hz,–CH2–NH2), 1.27 (s, 18H, C–CH3), 1.17 (s, 9H, C–CH3), 1.02 (s, 9H, C–CH3). IR: νmax (cm−1) 3391, 3274 (N–H), 1647 (CO), 1450 (C–N). MS–ESI calc. for C44H56O5N2S4 820.19, found 821.5 [M+H]+, 843.4 [M+Na]+, 1641.4[2M+H]+, 819.3 [M–H]−.
4.4.2 Synthesis of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(4-aminobutyl)aminocarbonyl)-methoxy]thiacalix[4]arene 3
5,11,17,23-tetra-tert-butyl-25-[(ethoxycarbonyl)methoxy]-2,8,14,20-tetrathiacalix[4]arene 1 (0.4 g, 0.49 mmol) and 1,4-diaminobutane (0.4 g, 4.5 mmol) were dissolved in 20 mL of THF. The yellow solution was refluxed for 24 h and 100 mL of a brine solution was added. The resulting precipitate was filtered off and taken up in CHCl3. The organic phase was extracted and washed with H2O (80 mL). The organic layer was dried over MgSO4 and the solvent was evaporated. The title compound was obtained as a yellowish powder (71%) with mp = 248 °C. 1H NMR (300 MHz, DMSO): 7.53 (d, 2H, Ar), 7.47 (d, 2H, Ar), 7.33 (s, 2H, Ar), 6.47 (s, 2H, Ar), 5.26 (s, 2H,–O–CH2), 3.22 (t, 2H, J = 7.15 Hz,–CH2–NH), 2.63 (t, 2H, J = 7 Hz,–CH2–NH2), 1.5 (m, 4H,–CH2), 1.25 (s, 18H, C–CH3), 1.11 (s, 9H, C–CH3), 1.04 (s, 9H, C–CH3). IR νmax (cm−1): 3295 (N–H), 1656 (CO), 1450 (C–N). MS–ESI m/z calc for C46H60O5N2S4 848, found 849.3 [M+H]+, 871 [M+Na]+, 1697 [2M+H]+, 847.4 [M–H]−, 1694.8 [2M–H]−
4.4.3 Synthesis of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(6-aminohexyl)aminocarbonyl)-methoxy]thiacalix[4]arene 4
5,11,17,23-Tetra-tert-butyl-25-[(ethoxycarbonyl)methoxy]-2,8,14,20-tetrathiacalix[4]arene 1 (0.7 g, 0.86 mmol) and 1,6-diaminohexane (0.6 g, 5.1 mmol) were dissolved in 20 mL of THF. The yellow solution was refluxed for 24 h and 100 mL of a brine solution was added. The resulting precipitate was filtered off and taken up in CHCl3. The organic phase was extracted and washed with H2O (80 mL). The organic layer was dried over MgSO4 and the solvent was evaporated. The product was obtained as a yellow solid (55%) with mp 260 °C. 1H NMR (300 MHz, DMSO): 7.53 (d, 2H, Ar), 7.48 (d, 2H, Ar), 7.33 (s, 2H, Ar), 6.4 (s, 2H, Ar), 5.3 (s, 2H, –O–CH2), 3.22 (t, 2H, J = 7.15 Hz, –CH2–NH), 2.74 (t, 2H, J = 7 Hz, –CH2–NH2), 1.51 (m, 4H, –CH2), 1.34 (m, 4H, –CH2), 1.25 (s, 18H, C–CH3), 1.11 (s, 9H, C–CH3), 1.03 (s, 9H, C–CH3). IR νmax (cm−1) 3390, 3276 (N–H), 1649 (CO), 1450 (C–N). MS–ESI m/z calc for C48H64O5N2S4 876, found 877.4 [M+H]+, 1753 [2M+H]+, 875.4 [M–H]−, 1751.9 [2M–H]−.
4.4.4 Synthesis of 5,11,17,23-tetra-tert-butyl-25-[2-imdazoline-methoxy]thiacalix[4]arene 5
4.4.4.1 Method A
A mixture of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(2-aminoethyl)aminocarbonyl)-methoxy]thiacalix[4]arene 2 (0.5 g, 0.24 mmol) and of 40 μL of triethylamine was heated at 170 °C during 30 min. The resulting layer was taken up in CHCl3. The organic phase was extracted and washed with H2O (40 mL). The organic layer was dried over MgSO4 and the solvent was evaporated. The product was obtained by precipitation from acetone.
4.4.4.2 Method B
A mixture of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(2-aminoethyl)aminocarbonyl)-methoxy]thiacalix[4]arene 2 (0.5 g, mmol) and of 40 μL of triethylamine was dissolved in 20 mL of CH2Cl2. The yellow solution was refluxed for 48 h. The organic phase was extracted and washed with H2O (80 mL). The organic layer was dried over MgSO4 and the solvent was evaporated. The product was obtained by precipitation from acetone.
The desired product (method A and B) was obtained by column chromatography on silica gel using chloroform/acetone (2:3 v/v) mixtures as eluents (20–25%) with mp = 272 °C. 1H NMR (300 MHz, CDCl3): 7.63 (m, 2H, Ar), 7.53 (m, 4H, Ar), 6.66 (s, 2H, Ar), 6.36 (s, 2H, –O–CH2), 4.12 (s, 4H, –CH2–CH2), 1.30 (s, 18H, C–CH3), 1.20 (s, 9H, C–CH3), 1.04 (s, 9H, C–CH3). IR νmax (cm−1): 3338 (N–H), 1450 (C–N). MS–ESI m/z calc. for C44H54O4N2S4 802 g/mol, found 803.4 [M+H]+, 1605.2 [2M+H]+, 801.6 [M–H]−,1603 [2M–H]−.
4.4.5 General procedure for the synthesis of bis thiacalix[4]arene 6–8
Under N2 atmosphere, a mixture of 5,11,17,23-tetra-tert-butyl-25-O-[(N-(2-aminoalkyl)aminocarbonyl)-methoxy]thiacalix[4]arene 2–4 (0.3 mmol), 4,4′-methylenebis(phenylisocyanate) (0.15 mmol) and two drops of dibutyltin dilaurate were stirred for 48 h at ambient temperature in 10 mL of dry THF. After distilling off the solvent by reduced pressure, the residue was purified by chromatographic column CHCl3/ethylacetate (4:1, v/v) as the eluent, then, compounds 6–8 were obtained as yellow powders.
4.4.5.1 4,4′-Bis[5,11,17,23-tetra-tert-butyl-25,26,27-trihydroxy-28-(phenylureylene-N-ethyl-carbamoyl-methoxy)thiacalix[4]arene]methylene 6
Mp = 205 °C. 1H NMR (300 MHz, DMSO): 7.58 (d, 6H, Ar), 7.54 (d, 4H, Ar), 7.44 (s, 4H, Ar), 7.28 (d, 4H, Ar), 7.03 (d, 4H, Ar), 6.8 (s, 2H, Ar), 6.03 (s, 4H, –O–CH2), 5.08 (s, 4H, –CO–CH2–NH), 3.73 (s, 2H, Ar–CH2–Ar), 3.15 (t, 4H, J = 7 Hz, –CH2–NH), 3.04 (t, 4H, J = 6 Hz, –CH2–NH), 1.31 (s, 18H, C–CH3), 1.23 (s, 36H, C–CH3), 1.14 (s, 18H, C–CH3). IR νmax (cm−1): 3250 (N–H), 1659 (CO), 1447 (C–N). MS–ESI m/z calc for C103H122N6O12S8 1892, found 1893.2 [M+H]+, 1890.4 [M–H]−.
4.4.5.2 4,4′-Bis[5,11,17,23-tetra-tert-butyl-25,26,27-trihydroxy-28-(phenylureylene-N-butyl-carbamoyl-methoxy)thiacalix[4]arene]methylene 7
Mp = 215 °C. 1H NMR (300 MHz, DMSO): 7.7 (d, 6H, Ar), 7.5 (d, 4H, Ar), 7.4 (s, 4H, Ar), 7.35 (d, 4H, Ar), 7.14 (d, 4H, Ar), 6.65(s, 2H, Ar), 6.0 (s, 4H, –O–CH2), 5.05 (s, 4H, –CO–CH2–NH), 3.72 (s, 2H, Ar–CH2–Ar), 3.2 (t, 4H, J = 7 Hz,–CH2–NH), 3.07 (t, 4H, J = 6 Hz, –CH2–NH), 1.32 (s, 18H, C–CH3), 1.20 (s, 36H, C–CH3), 1.10 (s, 18H, C–CH3), 0.9 (m, 4H, –CH2–), 0.59 (m, 4H–CH2). IR νmax (cm−1): 3254 (N–H), 1650 (CO), 1446 (C–N). MS–ESI m/z calc for C107H130N6O12S8 1947, found 1948.1 [M+H]+; 1946.5 [M–H]−.
4.4.5.3 4,4′-Bis[5,11,17,23-tetra-tert-butyl-25,26,27-trihydroxy-28-(phenylureylene-N-hexyl-carbamoyl-methoxy)thiacalix[4]arene]methylene 8
Mp = 219 °C. 1H NMR (300 MHz, DMSO): 7.67 (d, 6H, Ar), 7.58 (d, 4H, Ar), 7.46 (s, 4H, Ar), 7.31 (d, 4H, Ar), 7.20 (d, 4H, Ar), 6.7(s, 2H, Ar), 6.09 (s, 4H, –O–CH2), 5.06 (s, 4H, –CO–CH2–NH), 3.73 (s, 2H, Ar–CH2–Ar), 3.18 (t, 4H, J = 7 Hz, –CH2–NH), 3.07 (t, 4H, J = 6 Hz, –CH2–NH), 1.32 (s, 18H, C–CH3), 1.22 (s, 36H, C–CH3), 1.13 (s, 18H, C–CH3), 1.05 (m, 8H, –CH2–), 0.66 (m, 8H–CH2). IR νmax (cm−1): 3260 (N–H), 1640 (CO), 1444 (C-N). MS–ESI m/z calc for C111H138N6O12S8 2004, found 2005.7 [M+H]+.
Acknowledgments
The structure measurements were performed at the ‘Centre de diffractométrie Longchambon’ of the University of Lyon. The authors thank R. Vera for performing the X-ray diffraction analysis. We thank F. Albrieux, C. Duchamp, and N. Henriques from the Centre commun de spectrométrie de masse (ICBMS UMR 5246), for the assistance and access to the Mass Spectrometry facilities.