

1 Introduction
The improved performance and the low specific fuel consumption of diesel engines caused an increasing demand, during the last years, of cars powered by diesel engines. However, diesel engines produce NOx and particles of carbonaceous soot (PM), which consist of unburned organic compounds and other solid and liquid material. NOx and particulates from diesel engines have been identified to generate harmful effects on human health and the environment. To satisfy European and US regulations, extensive efforts have been focused on how to reduce emissions of pollutants, by controlling the combustion process and by developing efficient after-treatment systems. Currently, diesel particulate filters (DPF) are considered very effective a solution to attain the particulate matter (PM) emission standards since they have proved to meet serious diesel engine pollution reduction limits with filtration efficiencies exceeding 90 %. However, soot retained from exhaust gases should be removed to prevent back pressure and therefore a DPF regeneration is necessary. In the diesel exhaust emissions, NO2 and O2 are the main oxidants in presence. Hence, soot oxidation by these oxidants is an alternative to regenerate the filters. Several studies focused on the investigation of the uncatalyzed and catalyzed soot oxidation reaction by O2 and/or NO2 in the presence or absence of H2O. Many mathematical models were proposed to simulate these processes in order to understand thoroughly the inherent mechanisms, to predict the behaviour of DPF during its usage and to contribute to the improvement of the design process. Jeguirim et al. [1] studied the adsorption and reduction of NO2 at low temperatures (50 °C) on activated carbon and evidenced the formation of surface complexes such as –C(ONO2), –C(NO2) and –C(O). Gao et al. [2] found similar results. Muckenhuber and Grothe [3] proposed a reaction mechanism where two oxygen atoms from two different NO2 molecules are transferred onto the carbon surface. In this case, NO2 reacts directly with the carbon surface to form an acidic functional group, of acyl-nitrite type, as intermediate only. Du et al. [4] studied the oxidation by oxygen of uncatalyzed and calcium-catalyzed soot by means of Thermogravimetric Analysis (TGA) and Temperature-Programmed Desorption (TPD). They concluded that the products of the reaction, CO and CO2, are generated via different mechanisms and that CO2 was formed on sites different from CO ones. They formulated a model where the carbon structure is the controlling factor for the uncatalyzed oxidation and where calcium dispersion on the carbon surface is that for the catalyzed reaction. He et al. [5] simulated the CO/CO2 ratio obtained during char combustion by taking into account the pore model, the gas diffusion inside the pores and the reaction between carbon and oxygen. They concluded that the secondary reactions and pore structure significantly influenced the CO/CO2 ratio. Biggs and Agarwal [6] investigated the ratio CO/CO2 on a porous char particle in a fluidized bed and suggested a relationship between the CO/CO2 ratio and the char particle size. Floess et al. [7] found that the reactivity of char is a function of particle size for particles between 50 and 200 μm in diameter. This effect is not observed for macroporous char networks. Neeft et al. [8] studied the kinetics of the uncatalyzed oxidation, in oxygen/argon atmosphere with or without water, of two types of soot: flame soot (Printex U) and diesel soot, in the temperature range 450–550 °C in a flow reactor. A kinetic model, taking into account the conversion factor, was proposed and discussed. Jacquot et al. [9] and Jeguirim et al. [10–12] studied the kinetics of the reaction between NO2 and carbon in the presence of O2 and H2O in a fixed bed reactor. The rate increase of carbon consumption by NO2 in the presence of O2 was attributed to the reaction between NO2 and the intermediate species formed by the adsorption of oxygen on the carbon surface. Water presence increases the rate of carbon consumption because of the formation of intermediate nitric and nitrous acids which enhance the rate of C–NO2 reaction. However, the oxygen of water is not consumed and thus water is considered as a catalyst for the carbon oxidation reaction [12]. A monodimensional model was developed and kinetic parameters were extracted for the temperature range 300–400 °C [9–11]. Carbon oxidation by O2–NO2–H2O in a flow reactor was also studied at 250–500 °C by Jung et al. [13] who proposed reaction mechanisms and extracted kinetic constants. Schejbal et al. [14] developed a model for the soot deposition on the DPF and its regeneration based on the detailed kinetics of catalyzed and uncatalyzed soot combustion by O2 and NO2 developed by Jeguirim et al. [12]. The role of NO2 and O2 in the combustion of soot was also investigated by Setiabudi et al. [15] on three kinds of soot in the temperature range 100–450 °C in a flow reactor system and by thermogravimetry. The intermediates of soot oxidation were studied by infrared spectroscopy. Tighe et al. [16] studied the kinetics of oxidation by NO2 of three types of soot from a diesel engine in a packed bed at various temperatures (300–550 °C). The kinetics of the oxidation of four types of model and real diesel soot, by NO2 and O2, with or without water, in a flat bed reactor was also studied by Messerer et al. [17], who proposed a kinetic model to simulate experimental results. Kinetic data concerning the reaction between soot and NO2 have been also obtained by Kleffmann et al. [18], Arens et al. [19], Keil et al. [20], Prince et al. [21], Lur’e and Mikhno [22], Gray and Do [23] and Leistner et al. [24]. A theoretical study of the interaction between soot and NO in the absence of oxygen was carried out by Raj et al. [25] in order to develop the mechanistic understanding behind the formation of chemical species such as CO, N2 and N2O on soot. The energetics and kinetics were respectively evaluated using density functional theory and transition state theory. The model predicted well the formation of CO at temperatures > 600 °C using the rate observed experimentally in soot-NO environments. López-Fonseca et al. [26] established a kinetic model for the oxidation by oxygen of two diesel soot-like materials in a thermobalance (dynamic thermogravimetry). In the model established by these authors, the conversion factor was taken into consideration. Zouaoui et al. [27] proposed experimental and theoretical procedures to extract kinetic constants for the C–O2 reaction, taking into account oxygen diffusivity, from thermogravimetric experiments, in the temperature range 550–700 °C. Similar studies were carried out on soot and Printex U by Kalogirou and Samaras [28] and on two types of soot by Song et al. [29]. The catalytic combustion of carbon or soot by oxygen and/or NO2 has also received much attention [4,11,14,30–36]. Several kinetics data based on the catalytic mechanism were available.
The analysis of these literature data shows that most proposed models do not cover the whole temperature range 300–600 °C. Furthermore, there is a lack of data on the influence of the different components of diesel exhausts (O2, NO2, H2O) at different temperatures. It should also be added that the modification of the structure and physical properties of carbon or soot during the combustion process is well known in the literature but few simulation studies [4,7,8,16,17,26] took into consideration in their proposed models the variation of the kinetic rate of isothermal soot combustion with the conversion of soot mass, which is a consequence of the carbon structure variation. A recent investigation has used thermogravimetric analysis to propose a detailed set of kinetic reactions for soot oxidation by simulating diesel exhaust emissions but without investigating the effect of water vapour presence [37]. Hence, a detailed kinetic model for soot oxidation under real diesel engines conditions (NO2–O2–H2O) in a wide temperature range, 300–600 °C, where the influence of each gaseous species present is clearly taken into account and where the evolution of kinetic constants in isothermal conditions is considered, is necessary for car manufacturers and industrialists since simulation models can offer an important contribution to the improvement of the design process of diesel engines.
The objective of the present work is to perform experimental studies on carbon – taken as diesel soot model – combustion in conditions close to real diesel emissions, to study the influence of some main oxidants present in the real atmosphere of diesel engines (O2, NOx, H2O) at different temperatures (300–600 °C) and mixture compositions and then to elaborate a detailed kinetic model. Experimental studies will be carried out in a fixed bed reactor under a continuous flow of gases to mimic real diesel exhaust conditions.
2 Experimental part
The activity for soot oxidation was determined using commercially available carbon black powders Vulcan 6 (95.3 % C, 2.1 % O, 0.7 % H, 1 % S, and 0.3 % N). The use of this commercial soot for laboratory studies is chosen as it can be obtained in large quantities with reproducible characteristics unlike diesel soot.
Isothermal oxidation tests were carried out in a fixed-bed reactor (FBR) in a large range of temperatures (300–600 °C) and various oxygen, NOx and water concentrations (0–10 % O2, 0–600 ppm NO2, 0–10 % H2O). NO, very present in the diesel exhaust, is not discussed in this study since NO does not oxidize directly soot. Moreover, the role of NO on soot oxidation was only observed in the presence of catalyst and O2 [38]. The description of the FBR and the experimental procedure were reported elsewhere [9,10]. In each experiment, 10–50 mg of carbon black (CB) were used. The total flow rate was fixed to 100 NL·h−1 at 1 atm. The molar fractions of NO2, NO, CO2 and CO in the reactor exhaust were continuously measured by a UV absorption analyzer (Rosemount NGA 2000, Germany) and an infrared unit (MaihacMultor 610, France). Table 1 summarizes all the experimental conditions tested in this study.
Summary of our operating conditions.
| Temperatures (°C) | 300, 350, 400, 450, 500, 550, 600 |
| NO2 (ppm) | 200, 400, 600 |
| O2 (%) | 0, 2, 5, 10 |
| H2O (%) | 0, 2, 5,10 |
| Flow (NL·h−1) | 100 |
| Mass of CB (mg) | 10–25–50 |
The gas–solid reaction may occur in the diffusion and/or kinetic regime. To ensure that all the experiments reported here were not affected by such limitations, a series of experiments with various CB mass (10–50 mg, 100 mg and 200 mg) and flow rate (50 NL·h−1, 75 NL·h−1 and 100 NL·h−1) were previously examined [11,27]. No significant effect of the initial soot mass and of the flow rate on the specific rate of the soot oxidation was observed [11]. Moreover, it was checked that no significant exothermicity occurred during our isothermal runs. Heat limitations are negligible under our experimental conditions [27].
3 Results and discussions
Fig. 1 shows a typical temporal evolution of CO, CO2, NO and NO2 emissions in the FBR outlet during soot oxidation experiments (10 mg CB, 400 ppm NO2, 10 % O2, 5 % H2O, 500°C). Oxygen and water are not reported in the figure because they are in excess. For this reaction, emission curves of CO, CO2 and NO have similar shapes. These compounds are linked to the same oxidation mechanism.
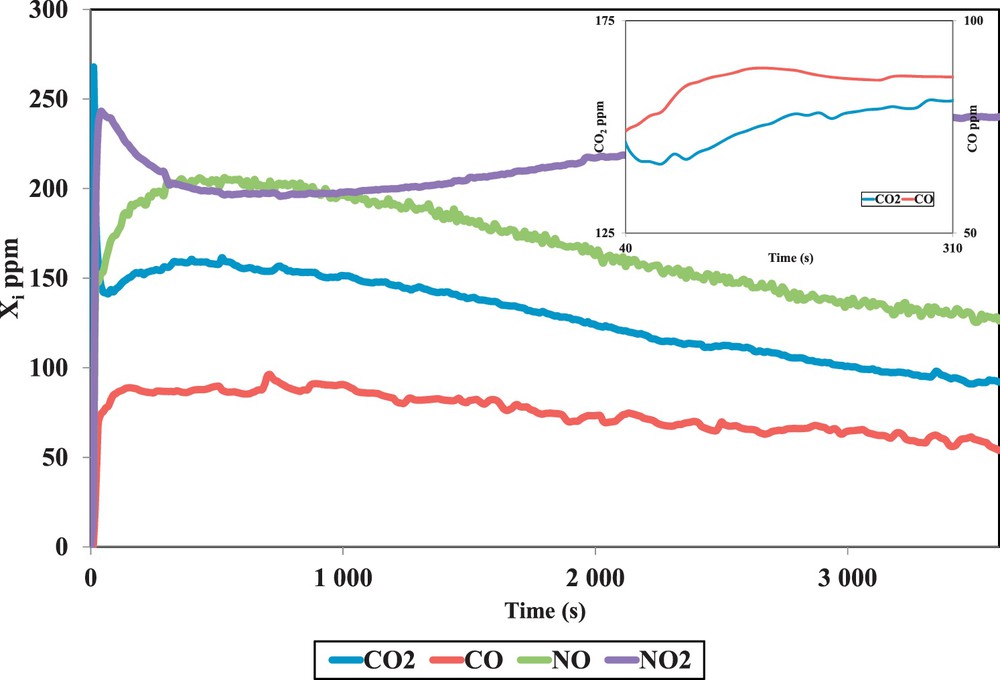
(Colour online) Outlet concentrations of CO2, CO, NO and NO2 versus time at 500 °C.
At the beginning of combustion, during 300 s, CO and CO2 concentration increases with time (Cf. the subfigure in Fig. 1) indicating an increase of the oxidation rate. The increase of the oxidation rate during the first 300 s is also observed for the various experimental conditions. In addition, the trend of CO and CO2 was similar during the first seconds for the different experimental tests and the ratio CO2/CO was almost constant for a conversion percentage lower than 10%. However, the CO2/CO values depend strongly on the gas inlet composition and temperature. This point is discussed further in the oxidation mechanism section.
After 300 s, CO and CO2 concentrations decrease with time. The increase of the combustion rate, during the first stage of combustion, was explained by several hypotheses. Jeguirim et al. attributed this first step to the formation of nitrogen species on the carbon surface [10]. Zouaoui et al. attributed the increase of the oxidation rate to an increase of the specific surface area of CB [27]. Indeed, they measured the specific surface area of CB, by the BET method, at different stages of CB combustion (for different conversion percentages). They found an increase in the specific surface area of the carbonaceous material with the increase of CB conversion up to 50 %.
Fig. 1 shows also that when the NO2 was turned on, it reacted with the soot, producing a peak in NO. The NO2 was also detected during the reaction, thus not all NO2 reacted with the soot. Nitrogen balance analysis confirms that NO and NO2 are the only nitrogenous species emitted during soot oxidation.
In order to identify the effect of operating conditions (gas composition, temperature), oxidation rates were compared at a fixed conversion percentage for the different experimental situations. The specific oxidation rate was calculated from the total gas flow rate and the CO and CO2 emissions using the following equation:
| (1) |
Fig. 2 shows the specific oxidation rate versus inlet gas composition and temperature for a soot conversion rate of 10 %. Fig. 2 shows that the direct oxidation of soot by NO2 starts at the low temperatures (∼ 300 °C). The oxidation rate is enhanced by the presence of O2 through the formation of C(O) complexes decomposed by NO2. Above 450 °C, the direct oxidation of soot by O2 starts and becomes the dominant reaction above 600 °C. Water vapour has a beneficial effect on the direct C–NO2 and C–O2 reactions. This beneficial effect decreases with temperature.
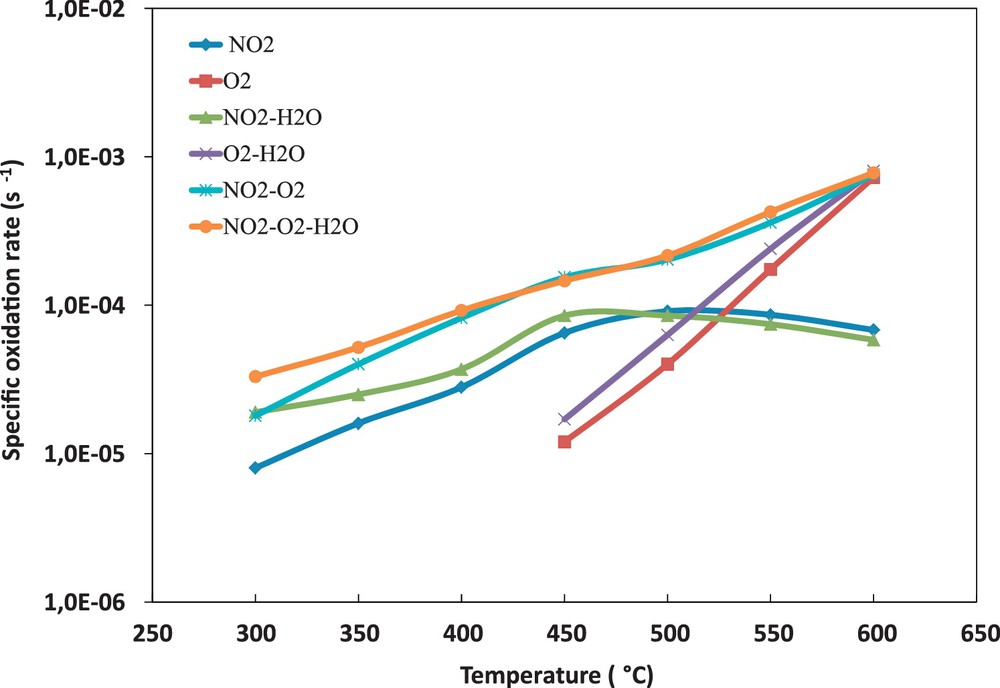
(Colour online) Influence of the inlet gas composition on the specific soot oxidation rate for 10 % soot conversion.
In order to assess further the role of each component on the reaction mechanism of soot oxidation, several calculations were performed. Hence, to get some information about the participation of H2O in the C–NO2 reaction, the oxygen contribution from oxygenated species NO2 and H2O was estimated [12]. These calculations show that the oxygen of water is not consumed and thus water is considered as a catalyst for the oxidation reaction of carbon [12]. This catalytic effect was attributed to the formation of intermediate nitric and nitrous acids which enhance the rate of C–NO2 reaction [10].
The beneficial effect of water vapour on the direct C–O2 reaction was also analysed. Hence, experimental tests of direct oxidation of carbon by water vapour were performed at the 450–600 °C temperature range. During, these tests, no significant oxidation of CB occurred. Therefore, the beneficial effect of water may be attributed to a catalytic effect. Such behaviour was mentioned previously by Ahlström and Odenbrand [43].
The evolution of the CO2/CO ratio was also examined for different experimental conditions. It was shown that the CO2/CO ratio decreases from 5 at 300 °C to 2.3 at 450 °C for the direct C–NO2 reaction. The values in the presence of water vapour are higher than those in the absence of water ranging from 6 to 2.7 between 300 and 450 °C.
The evolution of the CO2/CO ratio during the direct C–O2 reaction in the absence and in the presence of water vapour was assessed. It was observed that the CO2/CO ratio decreases from 1.6 to 0.8 in the absence of water and from 2 to 0.95 in the presence of water between 500 and 600 °C for a conversion percentage of 20 %. It was also observed that the CO2/CO ratio decreases in the absence of water with time until a conversion percentage of 50 %. In contrast, the CO2/CO ratio increases in the presence of water with time until a conversion percentage of 50 %. These observations may confirm the beneficial effect of water on the direct C–O2 reaction between 500 and 600 °C.
The CO2/CO ratio during the C–NO2–O2 reaction was also evaluated. The obtained values were higher in the presence of water (5.1 at 300 °C to 1.1 at 600 °C) compared to those obtained in the absence of water (4 at 300 °C to 0.9) at 600 °C.
In order to develop the kinetic model, the influence of the main oxidant concentration was also performed. Hence, it was observed that the rate of carbon consumption as well as the rates of CO and CO2 formation increases linearly with the increase of the NO2 inlet mole fraction at a given temperature. Such results prove that the reaction order with respect to NO2 may be close to one.
In addition, for a fixed NO2 mole fraction (∼ 400 ppm), the increase of O2 concentration, at two different temperatures, 300 °C and 400 °C, resulted in an increase of the rate of carbon consumption. These results are taken into account to determine the reaction order with respect to O2
Furthermore, for a NO2 mole fraction of ∼ 400 ppm and 5 % O2, at 300 °C, the increase of the water vapour inlet mole fraction (0 to 10 %) leads to the increase of the rate of carbon consumption. These experiments are used to determine the reaction order with respect to H2O. The obtained results in the previous and current investigations show that the oxidation mechanism of carbon by NO2 and O2 comprises two main simultaneous reactions [9–11]:
- • a direct reaction between carbon and NO2 or O2:
(2) (3) (4) (5) - • a cooperative reaction involving simultaneously NO2 and O2:
| (6) |
| (7) |
In the above mechanism, it is not assumed that CO2 could be obtained by CO oxidation by O2 since some authors [4] found that, during soot combustion, CO2 is formed on different sites than those of CO.
In addition, it was proven that H2O exerts a catalytic effect on the direct oxidation of carbon by NO2 [9–11]. A beneficial effect of H2O on the direct reaction between carbon and O2 is also observed in the present study. Both H2O catalytic effects on C–NO2 and C–O2 reactions are taken into account in this work.
Recent investigations of soot oxidation by simulated diesel exhaust emissions using thermogravimetric analysis confirmed the proposed mechanism [37]. In fact, Lee et al. have noted a lower temperature zone for soot oxidation ranging from 288 to 500 °C and a higher temperature zone ranging from 516 to 626 °C [37].
3.1 Kinetics of Soot-NO2 reaction
From Eqs. (2) and (3), the following equation has been used to derive the kinetic parameters of soot oxidation:
| (8) |
The dependence of the different intrinsic rate constants on temperature is expressed by the Arrhenius function (s−1):
| (9) |
The values of A and Ea are extracted using the previously developed monodimensional model [9–11] of soot oxidation, in which consumption of NO2 through the soot bed was taken into account. Therefore, the soot bed is split into small layers and the specific oxidation rate is determined by computing for each elementary layer of the fixed bed, the specific oxidation rate and the depletion of NO2. The specific oxidation rate was obtained by summing the different rates in each layer and comparing this value with the experimental one. The main features of the modelling procedure are given in previous investigations [9–11].
Experiments in the temperature range of 300–600 °C allowed us to describe the temperature dependence of kinetic constants
| (10) |
| (11) |
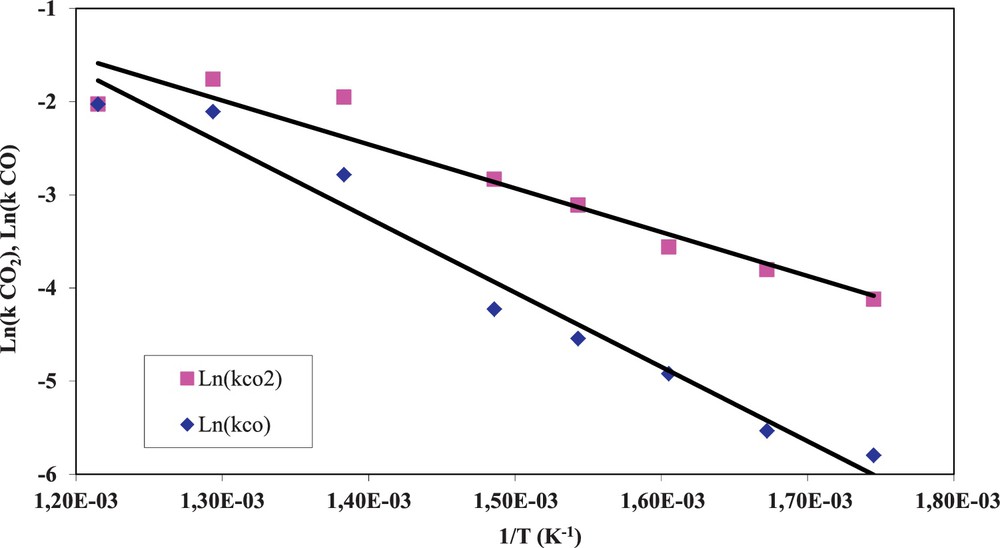
(Colour online) Arrhenius plot of the kinetic constants of soot-NO2 reaction.
The obtained values for activation energies for soot–NO2 reactions were not affected by the conversion rate. These values are very close to the activation energies reported by other authors for the oxidation of soot by NO2. In particular, Lur’e and Mikhno mentioned an activation energy of 50 kJ·mol−1 for graphitized soot oxidation by
3.2 Kinetics of soot–NO2–H2O reaction
Although water vapour is a major component in the automotive exhaust gas, its effect is not well examined in the literature. Previous investigations have mentioned that water vapour exerts a catalytic effect on the oxidation of soot by NO2 in the presence of oxygen [9]. Jeguirim et al. have proved that the catalytic effect of water affects only the direct C–NO2 reaction – Eqs. (2) and (3) – and does not affect the cooperative reaction – Eqs. (6) and (7) – [10]. The catalytic effect of H2O is attributed to the intermediate formation of traces of nitric and nitrous acids, which enhance the rate of carbon oxidation without modifying the global reaction mechanism [12]. The present study shows that the effect of water on the direct C–NO2 reaction decreases when temperature increases. Such a behaviour should be taken into consideration for the developed kinetic model. Hence, additional terms (
| (12) |
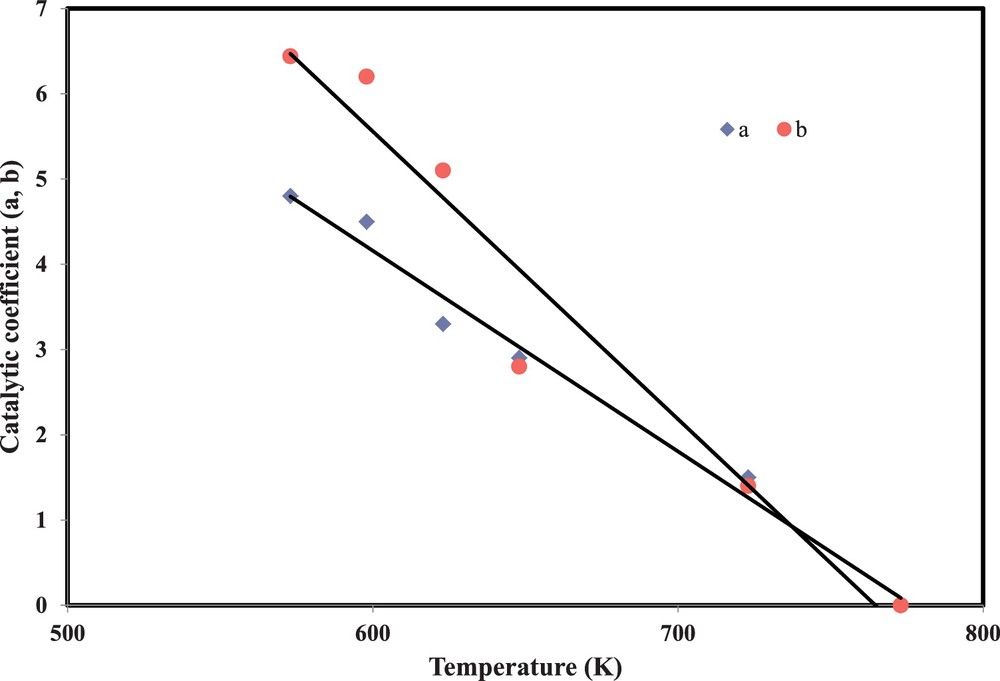
(Colour online) Evolution of water catalytic coefficients with temperature.
The best fit was obtained for:
| (13) |
| (14) |
α = 0.4
β = 0.6
3.3 Kinetics of soot–O2 reaction
The oxidation of soot by oxygen starts at a much higher temperature, around 450 °C (Fig. 2), compared to NO2, and becomes very fast at 600 °C. Several papers have examined the direct oxidation of soot by oxygen [8,40]. This oxidation is characterized by a direct reaction between oxygen and carbon:
| (4) |
| (5) |
As for NO2, the oxidation rate rdirO2 can be written as:
| (15) |
Experiments in the temperature range of 450 to 600 °C, those of soot oxidation by oxygen, allowed us to describe the temperature dependence of kinetic constants
| (16) |
| (17) |
The obtained activation energy values are quite close to the ones reported by other researchers: Neeft et al. calculated Ea = 168 kJ·mol−1 for the oxidation of soot by 10 % O2 in a temperature range of 442–527 °C [8]. Yezerets et al. reported Ea = 137 kJ·mol−1 for soot oxidation with an oxygen concentration of 3–25 % and temperatures ranging from 400 to 550 °C [39,40]. Recently, Lee et al. reported an activation energy close to 155 kJ·mol−1 for the soot oxidation by simulated diesel exhaust emissions in the temperature range 516–626 °C [37]. Wang–Hansen et al. mentioned an activation energy of 157 kJ·mol−1 during their kinetic analysis of O2-based oxidation of synthetic soot [42]. Leistner et al. obtained Ea = 164 kJ·mol−1 for the reaction leading to the emission of CO – Eq. (4) – and Ea = 147 kJ·mol−1 for the reaction leading to the emission of CO2 – Eq. (5) – [24].
3.4 Kinetics of soot–O2–H2O reaction
Experiments of soot oxidation by oxygen performed in the presence of water show a beneficial effect of the presence of water on the oxidation rate. Such a behaviour was previously observed by Ahlström and Odenbrand during the investigation of the combustion characteristics of soot deposits from diesel-powered engines in the presence of 2–10 % O2 and 0 or 7 % H2O [43]. Such a behaviour should be taken into account for the developed kinetic model. Hence, the additional term (
| (18) |
c(T) = 0.3
γ = 0.2
3.5 Kinetics of soot–NO2–O2 reaction
During the oxidation of soot by NO2 and O2 between 300 and 600 °C, three reactions occur simultaneously: the direct oxidation of soot by NO2, the direct oxidation of soot by O2 and the cooperative reaction involving a synergetic effect of oxygen and NO2. However, the contribution of each reaction on the global oxidation rate depends on temperature. Hence, the following equation may be used to derive the kinetic parameters of the oxidation of soot by NO2 and O2:
| (19) |
| (20) |
| (21) |
| (22) |
The kinetic parameters for the cooperative reaction were obtained from the experiments of soot oxidation by NO2 and O2 taking into account the occurrence of the direct C–NO2 and direct C–O2 reactions. The cooperative reaction rate was calculated by subtracting from the global oxidation rate (r) the rate of the direct reaction C–NO2 (
| (23) |
| (24) |
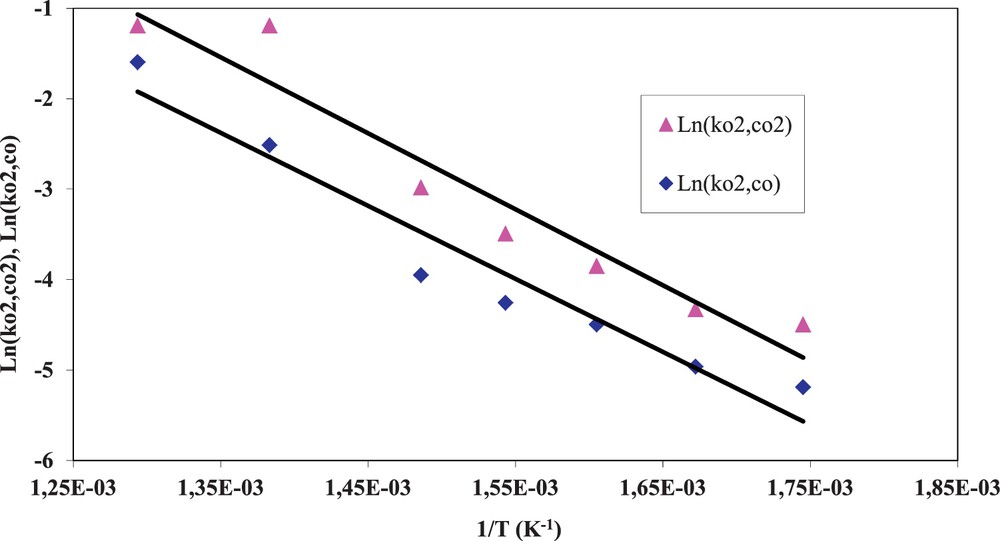
(Colour online) Arrhenius plot of the kinetic constants for the cooperative reactions.
The obtained activation energy values are very close to the values (60–80 kJ·mol−1) defined by Messerer et al. for the overall process of adsorption and reaction during the oxidation of soot by NO2 and O2 [17]. Leistner et al. obtained 70 kJ·mol−1 for the reaction involving the decomposition of C(ONO2) complexes into CO2 and NO [24].
3.6 Kinetics of soot-NO2–O2–H2O reaction
During the oxidation of soot by a mixture containing oxygen, nitrogen dioxide and water vapour, it was shown that the direct C–NO2 and C–O2 reactions and the cooperative C–NO2–O2 reaction occur simultaneously. Water vapour has a catalytic effect on both direct reactions but does not significantly influence the cooperative reaction (Fig. 2). Hence, the global oxidation rate of soot oxidation by diesel exhaust emission may be expressed by:
| (25) |
The obtained values of the different kinetic parameters were determined previously for each reaction and are summarized in Table 2. Using the obtained kinetic constants, the intrinsic soot oxidation rates for the different reactions are calculated for a gas mixture containing 400 ppm NO2, 10 % O2 and 5 % H2O at a temperature range from 300 to 600 °C. The obtained values are compared in Fig. 6.
Kinetic parameters for soot oxidation by NO2, O2 and H2O.
| α | 0.4 |
| β | 0.6 |
| γ | 0.2 |
| a(T); T(K) | 18.25 – 0.024 T |
| b(T); T(K) | 25.92 – 0.034 T |
| c(T) | 0.3 |
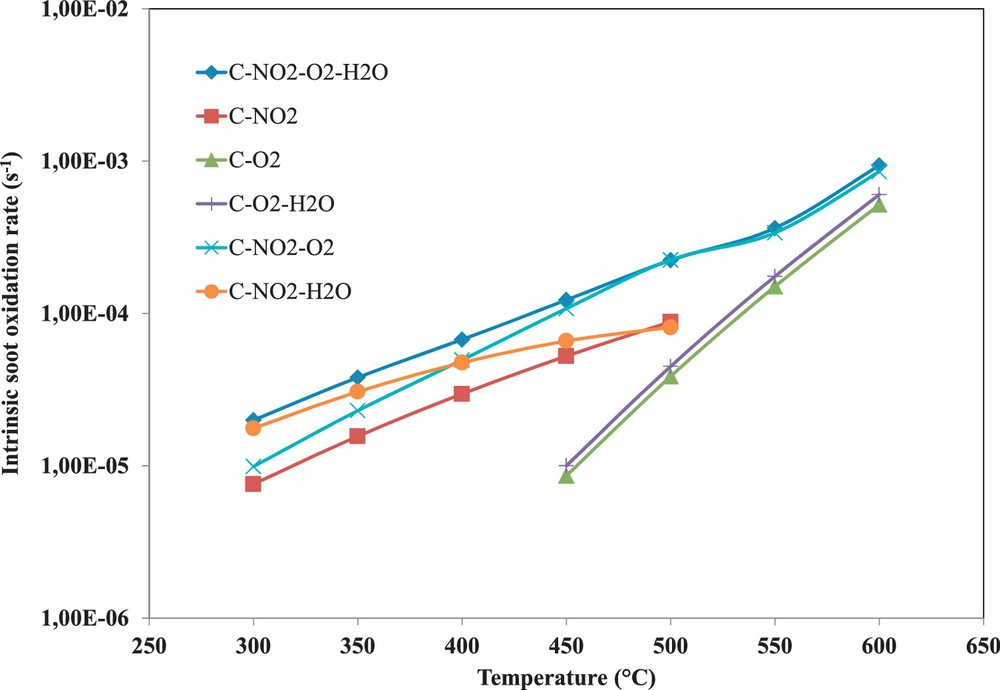
(Colour online) Comparison between the calculated intrinsic oxidation rates of soot oxidation.
Fig. 6 shows that the calculated intrinsic oxidation rates confirm the role of each component from the exhaust gas on the mechanism of soot oxidation in diesel exhaust emissions. Hence, from 300 to 450 °C, the direct oxidation of soot by NO2 and the cooperative C–NO2–O2 reaction are responsible for soot oxidation. In this temperature range, water vapour has a catalytic effect on the direct C–NO2. Above 450 °C, the direct oxidation of soot by O2 starts and becomes the dominant reaction above 600 °C. The addition of water vapour leads to a slight increase of the oxidation rate of soot by oxygen.
The above equations were also verified for the different inlet NO2, O2 and H2O concentrations used. The as-obtained calculated rates of soot consumption were in good agreement with the experimental ones. This agreement tends to be better as the rate of consumption decreases. However, the mean standard deviation is equal to 10 %.
4 Conclusion
Carbon black was used as a model for diesel soot and was oxidized under modern diesel engine emission conditions, 300–600 °C, NO2 (0–400 ppm), O2 (0–10 %) and H2O (0–10 %), in a fixed bed reactor. The gases issued from combustion were exclusively CO, CO2 and NO.
It was found that carbon is mainly oxidised by NO2 at 300–450 °C and by O2 at 450–600 °C, and that water has a catalytic effect on these reactions, called direct reactions. Moreover, in the presence of both NO2 and O2, the oxidation rate was enhanced, compared to both direct reactions, without the catalytic effect of water.
According to experimental data, a kinetic mechanism was established and then a mathematical one-dimensional fixed bed model was proposed. Kinetic constants for each identified reaction were determined by the data fitting of the theoretical CO and CO2 emission curves with the experimental ones. The kinetic constants have shown an Arrhenius behaviour. Moreover, constants related to the water catalytic effect were also determined by modelling.
The comparison between the experimental oxidation rates and the ones obtained by the kinetic constants extracted by modelling shows a good agreement between the experimental data and the kinetic mechanism proposed.
This study contributes to the investigation of the oxidation mechanism of diesel soot in real conditions. Some further investigations should be done, such as the influence of mass carbon conversion on the overall combustion process, before proposing a set of kinetic constants to car manufacturers in order for them to predict the behaviour of the diesel filter particulate regeneration.
Acknowledgements
M. Labaki would like to thank the “Agence universitaire de la Francophonie (AUF) – Région du Moyen–Orient”, for financial support. The authors are also grateful to Mrs Damaris Kehrli and Nelly Dawson for carrying out some experiments. The authors would like to thank the Laboratoire “Gestion des risques et environnement” for technical support.