

1 Introduction
As diesel engines have improved in fuel efficiency, sales of diesel cars have greatly increased over the last 20 years to the detriment of gasoline vehicles. However, diesel engine exhaust contains more soot and NOx than gasoline vehicle exhaust. Because of the toxicity of soot and NOx to the environment and to human health, emissions of these components are strictly limited according to emissions standards. For this reason, converters after treatment are necessary for diesel cars.
For NOx treatment, two different processes are used: selective catalytic reduction (SCR) with an injection of ammonia or urea and NOx storage reduction (NSR), which needs lean-rich burn cycles. The latter technology is more suited for passenger cars and makes no use of an external reductant. The lean-rich burn cycles consist of alternating lean periods during which NOx is stored as nitrate on the catalyst [1–3] and shorter rich periods in which stored nitrates desorb and are reduced to N2 [4–7].
The NSR catalysts contain two major components: alkaline/alkaline-earth metal oxides (K2O and/or BaO), which act as the NOx storage materials, and precious metals, such as Pt, Pd and Rh, which allow NO oxidation during the lean phase and NOx reduction during the rich period. The active phase is supported on a high-surface-area support, such as γ-Al2O3. The behavior of NSR catalysts or model NSR catalysts, such as Pt–Ba/Al2O3, Pt-K/Al2O3 or Pt-K-Ba/Al2O3, is well documented in the literature. In the last decade, several papers have focused on a better understanding of the NOx storage mechanism [1,8–12], the effect of the composition of reactive gas on both NOx storage capacity [1,13,14] and the adsorption mechanism [1,9,13,15–20]. Several authors [1,13,15,21,22] agree that the presence of CO2 or H2O in the gas flow induces a decrease in the NOx adsorption capacity (NSC) of the NSR catalysts. However, the influence of these gaseous compounds is different, as water particularly affects the NSC at low temperatures (< 300 °C), while the effect of CO2 is observed at higher ones [1,13]. Although studies presented in the literature agree that the decrease in NSC observed in the presence of CO2 in the gas flow leads to a competition between the formation of nitrate and the formation of carbonate, the role of water in the NOx adsorption mechanism is still under debate. Recent studies [12,15,20,22] proposed that water particularly affects the oxidation of NO into NO2 over Pt sites. These phenomena would be partially irreversible. The decrease in the NSC observed in the presence of H2O in the gas flow is thus attributed to a decrease in the local NO2 concentration. Corbos et al. [1] proposed that water particularly affects the adsorption of NOx on alumina sites. In our previous work [22], we proposed that the formation of nitrate bulk species is not affected by the presence of water, while the oxidation of bidentate nitrites into bidentate nitrate surface species is slowing down, leading to the preferential formation of monodentate nitrate surface species.
To control soot emissions, diesel particulate filters (DPFs) are typically used [23,24]. The filters should periodically be regenerated to eliminate entrapped soot and to prevent an excessive pressure drop. The presence of NOx, especially NO2 in the gas flow, is known to facilitate soot oxidation by O2 [10,25–27]. In the average temperature range of diesel exhaust (300 °C–400 °C), NO2 allows the destabilization of surface oxygen complexes, such as C(O), and then the oxidation of soot [10,27]. With a gas stream composed of a mixture of NO2 and O2, continuous soot combustion in the filter can be achieved [10,25,28]. The presence of a Pt catalyst has been reported to increase the surface concentration of C(O) complexes, which then react with NO2, leading to enhanced carbon consumption [29]. Jeguirim at al. reported that a Pt-based catalyst has no effect on the direct oxidation of carbon by O2 or NO2, but in the presence of a NO2O2 mixture such as reactive gas, Pt exerts a catalytic effect on the cooperative CNO2O2 oxidation reaction [30]. Injection of water vapor was also observed to assist soot oxidation without changing the global mechanism, in which the water acts as a catalyst for the CNO2 reaction [26,28].
A treatment system for diesel engines that combined these two after-treatment devices (NSR and DPF) was developed by Toyota and called Diesel Particulate NOx Reduction (DPNR) [31,32]. DPNR technology is based on the use of a particulate wall-flow filter coated with an NSR catalyst layer working under cyclic conditions. This system, named a “four-way catalytic converter”, has the capacity to remove volatile organic compounds (VOCs), CO, NOx and soot simultaneously. Particulate filter regeneration is effective during the lean phase due to the presence of NOx and an excess of oxygen in the exhaust gas, similar to CRT technology. Soot oxidation is also claimed to occur during the rich phase [31].
The combination of NSR and continuously regenerating trap (CRT) technologies impacts their performance so that interactions between soot removal and NOx removal functions appear. Recent studies have been conducted to highlight these interactions, their consequences on the reactivity of NSR catalysts and the kinetics of soot combustion [33–40]. Although most of these investigations were focused on the influence of the presence of a model NSR catalyst (Pt–Ba/Al2O3 or Pt–K/Al2O3) on soot oxidation, some authors [33,35,38,40,41] investigated the influence of soot-NSR catalyst interaction on the NOx storage mechanism. These recent studies agree that the presence of soot induces a decrease in the NSC. In agreement with Artioli et al. [38], previous work from our group [35,40] led us to conclude that nitrate species stored on Ba or Al sites in contact with soot are destabilized according to the following direct surface reaction between carbon particles and NOx storage sites to form adsorbed carbonate species:
| (1) |
| (2) |
Our results showed that carbon contact with the NOx storage catalyst affects nitrate species formed by the “nitrate route” (far from Pt sites) more than the nitrate species formed by the “nitrite route”. Moreover, we observed that the carbon oxidation process leads to premature aging of the catalysts and induces structure modifications (Pt sintering and Ba agglomeration). These structural modifications reduce the proximity between Pt and the adsorption sites (Ba and/or Al), which results in a decrease of NOx storage through the “nitrite route”.
Based on these findings, in the present work, we investigated the influence of the reactive gas composition, and more precisely, the effect of the presence of H2O on the catalytic performance of a model four-way catalyst (1% Pt–10% BaO/Al2O3 in contact with carbon black). Indeed, as mentioned above, H2O inhibits the NOx storage on NSR catalysts. However, studies that investigated the interactions between soot and NSR catalysts were always performed under constant conditions. For this purpose, using well-selected testing procedures in a fixed-bed reactor and also in situ DRIFTS, XRD and XPS characterization, we investigated to what extent contact between carbon and the NSR catalyst influences the impact of water.
2 Experimental
2.1 Catalyst preparation
A Pt/Ba/Al2O3 NSR catalyst (1/10/89 w/w) was prepared by incipient wetness impregnation. γ-Alumina pellets were crushed in order to obtain particles with a diameter between 250 and 400 μm. Alumina (Alfa Aesar, 220 m2/g) was impregnated by a platinum solution (Pt(NH3)2(NO2)2; Aldrich Chemistry, 3.4% Pt in ammonium hydroxide) under agitation and heating until the solvent was evaporated, then dried at 110 °C for a night and calcined in air at 500 °C for 2.5 h. The support was impregnated with an aqueous solution of barium nitrate (Fluka, 99% purity) and dried overnight at 120 °C before being calcined at 500 °C for 2.5 h in the air in order to obtain the catalyst. The catalyst was then sieved in the 250–400-μm range. The loadings of Pt and Ba were 1 wt% and 10 wt%, respectively.
A commercial carbon black (CB), Vulcan 6 from Cabot with a specific surface area equal to 106 m2 g−1, was used as a model for soot particles. Before to be mixed with the catalyst, CB was crushed in a mortar for 10 min then sieved in the 30–50-μm range.
2.2 NOx storage measurement
The catalytic activity for NOx adsorption and carbon oxidation was measured using a fixed-bed quartz reactor (internal diameter: 16 mm) placed in the experimental set up described in reference [35]. Catalytic tests were performed either with pure catalyst or in the presence of a carbon–catalyst mixture. Catalytic tests with the pure catalyst were carried out on a 656-mg sample in the reactor. For the experiments performed in the presence of the carbon, CB was mixed in a mortar with the catalyst in a 1: 6 w/w ratio during 15 min. A tight contact was preferred in this work in order to enhance the effect of carbon/catalyst interaction as compared to loose contact.
The NOx storage capacity of the catalyst was studied by performing isothermal NOx adsorption/temperature-programmed desorption (TPD) cycles in a vertical flow quartz reactor described in [35]. The adsorption phase was performed at 300 °C during 1 h with a reactive gas mixture containing 300 ppmv NO, 10% b.v. O2, 0–5% bv H2O in N2. Then the reactive gas was switched to pure N2 and the temperature was cooled to 423 K. Subsequently, TPD of stored NOx was carried out by heating the sample at a rate of 5 K/min under pure N2 from 423 to 873 K. The samples (catalyst or carbon–catalyst mixture) have been conditioned by performing four NOx adsorption/TPD cycles in order to obtain a reproducible behavior. Note that each NOx adsorption/TPD cycle was separated by the reducing surface treatment under 2% H2 in N2 at 573 K during 30 min. All the gases were injected via different mass flow meters except H2O. The water vapor was introduced in a preheated gas mixer before to be injected in the reactor. The outlet gases were analyzed by a multi-component gas analyzer (Rosemount NGA2000).
2.3 Catalyst characterizations
Diffuse reflectance (DRIFTS) spectra were recorded in the 4000–700-cm−1 range (resolution 4 cm−1, 100 scans) with a Varian Excalibur 4100 FTIR spectrometer equipped with a MCT detector, a “Graseby Specac” optical accessory and a Spectra-Tech environmental cell. Prior to adsorption tests, the NSR catalyst was pre-treated in situ under He at 693 K. Then the temperature was cooled down to 573 K in He and a background spectrum was recorded. NO adsorption tests were carried out at 573 K using two different gas mixtures:
- • NO 1500 ppm, O2 10 bv%, He balanced;
- • NO 1500 ppm, O2 10 bv%, H2O 1 bv%, He balanced. DRIFTS spectra were monitored according to time up to saturation of the surface by adsorbed species.
XRD patterns were recorded on a Bruker D8 powder diffraction system using a Cu Kα radiation source equipped with a graphite monochromator. The diffraction patterns were collected under ambient conditions over the 10–90° range. The phases were identified by comparison of the measured set of interplanar distances, d, and of the corresponding intensities of the diffraction maximum, I, with those found in the JCPDS database. Transmission electron microscopy (TEM) images were obtained with a TopCon 2100 FCs microscope operated at 200 kV. XPS spectra were recorded between 200 and 800 cm−1 and are presented as normalized spectra. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Multilab 2000 spectrometer equipped with an Al Kα X-ray source (1486.6 eV). The XPS analyses were performed in a static system; the base pressure during the analysis was 10−9 Pa. Binding energies were corrected for charge effects by referencing to the C1s peak at 284.6 eV. All samples characterized by XDR, XPS and TEM, except γ-Al2O3, are conditioned samples, removed from the reactor after an adsorption phase performed after the four adsorption/TPD cycles. So that, based on the NOx storage capacity study (§3.5), these surfaces are assumed to be saturated in NOx.
3 Results and discussion
3.1 XRD measurements
The XRD patterns for the γ-Al2O3 support are presented in Fig. 1a, and are compared to pure catalyst or carbon–catalyst samples conditioned under different treatments; i.e. adsorption/TPD cycles performed in the presence (Fig. 1b and c) or the absence (Fig. 1d and e) of water. The general profile for the diffraction peaks reveals the nanocrystalline character of the samples. All of the catalyst and carbon–catalyst samples consisted of γ-Al2O3 with broad peaks and nearly the same intensity, but largely, their peak intensities at 2θ = 66.73° and 45.95° were less than those in the γ-Al2O3 support.
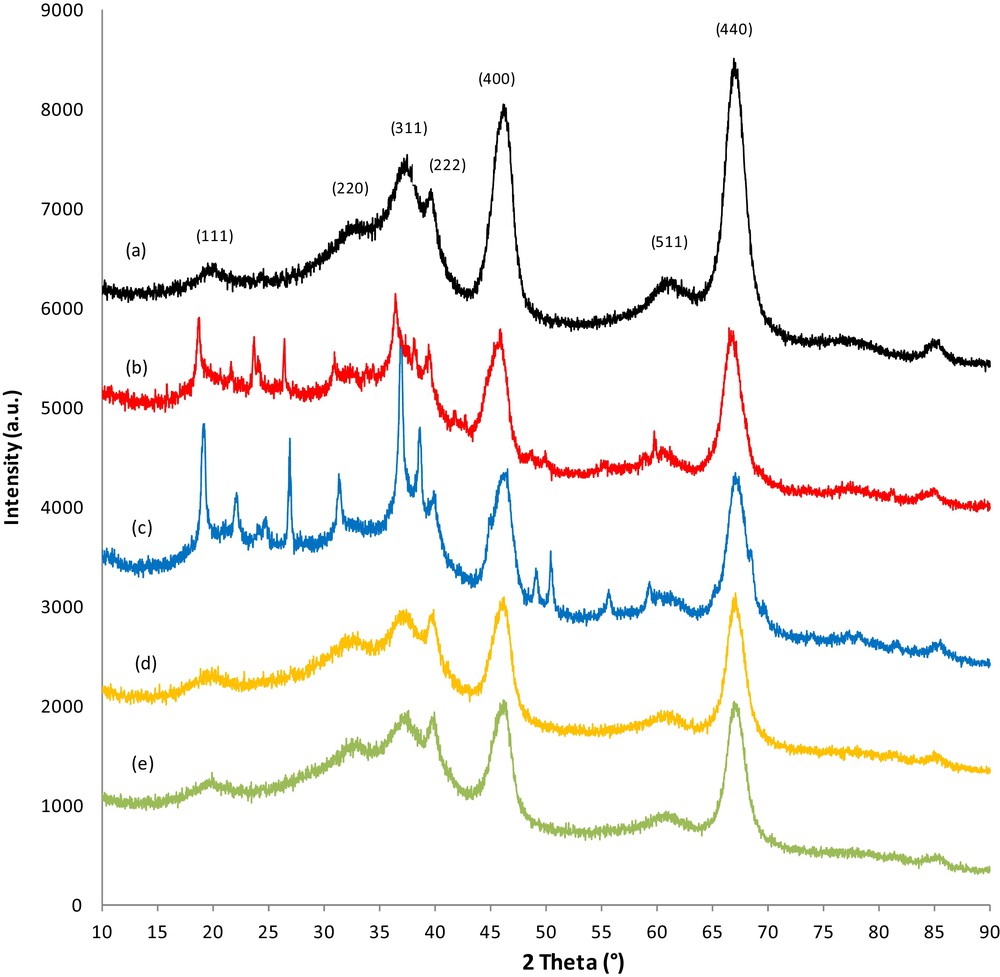
(Color online). X-Ray diffractograms obtained with γ-Al2O3 (a), a carbon–catalyst sample conditioned in the presence of water in the reactive flow (b), a pure catalyst sample conditioned in the presence of water in the reactive flow (c), a carbon–catalyst sample conditioned without water in the reactive flow (d), and a pure catalyst sample conditioned without water in the reactive flow (e).
For all catalyst samples, the diffraction peaks were observed at 2θ = 18.86°, 31.58°, 36.97°, 39.17°, 45.95°, 60.05, and 66.73°. These reflections are attributable to the γ-Al2O3 structure and correspond to the Miller indices of (111), (220), (311), (322), (400), (511), (440), respectively. The patterns were found to match the standard data of γ-Al2O3 (JCPDS 75-0921). The lattice parameter value of γ-Al2O3 is approximately 0.7927 nm in all catalyst samples, which appears to be independent of the catalyst's treatment. The average crystallite size of γ-Al2O3 samples estimated by the Scherrer equation is 35 nm.
Comparing the diffractograms of the samples after adsorption/TPD cycles without water (Fig. 1d and e) to that of the alumina support, we observed that neither conditioning nor soot treatment altered the structure of the γ-Al2O3 support. It is worth noting that no peaks were attributable to Pt, BaO, BaCO3, or BaNO3 and that no other impurities were detected, which indicates an interaction between the Ba and Pt phases under our conditions and/or that Ba species are present in (an) amorphous phase(s). The absence of diffraction peaks corresponding to Pt can be explained by the following reasons:
- • either the loading of Pt is low;
- • Pt is finely dispersed.
This result is reasonable because the catalysts were calcined at 500 °C before the reduction treatment, which, in this case, impeded the aggregation of Pt particles. Similar results regarding the positive influence of high calcination temperatures prior to the reduction treatment have been observed in other research studies [42–44].
Introduction of water (Fig. 1b) during the adsorption/TPD cycles induces some structural changes in the structure of the catalyst. Together, the peaks corresponded to γ-Al2O3, and other peaks were observed at 2θ = 21.99°, 24.59°, 26.74°, 31.28°. These reflection peaks can be attributed to Ba(NO3)2, suggesting that the presence of water either reduces the interaction between Pt and Ba and/or acts as a mineralizing agent which allows NOx to be in an ionic form on the surface of barium. Thus the presence of water seems to favor the formation of a crystalline phase instead of an amorphous form (pseudomorphism). When soot is present in the catalytic bed (Fig. 1c), the reflection peaks corresponding to Ba(NO3)2 are observed yet. However, their intensity is decreased. It is worth nothing that the reflection peak at 2θ = 26.88° in the samples with soot can also be attributed to BaCO3. However, no diffraction peaks corresponding to Pt were observed in any of the samples. This result was most likely due to the low Pt content and should be covered by Ba or due to oversampling of the signal in most cases. These results could suggest also a high amount of Pt dispersion.
3.2 XPS analysis
XPS analysis is performed to elucidate the surface chemical composition and the oxidation state on the surface for the catalysts. XPS survey scan analysis is carried out in a binding energy range between 0 and 850 eV (Fig. 2). The survey spectrum shows that Al, Pt, Ba, C, and O are present on the surface and that there are no significant impurities present, except for the contaminant carbon in the carbon–catalyst mixtures and the adventitious hydrocarbon in the XPS instrument itself. The XPS binding energies of these elements are tabulated along with the standard values [45]. The binding energies of the peaks (after the corrected spectrum) and their assignments are reported in Table 1.

XPS survey scans obtained with a pure catalyst sample conditioned in the presence of water in the reactive flow (a), a carbon–catalyst sample conditioned in the presence of water in the reactive flow (b), a pure catalyst sample conditioned without water in the reactive flow (c), and a carbon–catalyst sample conditioned without water in the reactive flow (d).
XPS binding energies.
| Sample | Conditioning | Binding energy (eV) | ||||
| Al 2p | C 1s | Pt 4d5/2 | O 1s | Ba 3d5/2 | ||
| Pure | With water | 74.1 | 284.6 (100%) | 314.4 | 529.9 (100%) | 779.9 (100%) |
| Carbon–catalyst | With water | 74.1 | 284.6 (56%) | 314.7 | 529.9 (87%) | 779.8 (92%) |
| 288.5 (33%) | 532.9 (13%) | 780.4 (8%) | ||||
| 293 (11%) | ||||||
| Pure | Without water | 74.1 | 284.6 (70%) | 314.4 | 530.1 (100%) | 779.8 (95%) |
| 292 (30%) | 780.3 (5%) | |||||
| Carbon–catalyst | Without water | 74.1 | 284.6 (50%) | 314.7 | 529.9 (89%) | 779.7 (94%) |
| 288.7 (29%) | 532.7 (11%) | 780.3 (6%) | ||||
| 292.8 (21%) |
According to the literature [46], the binding energy of Al 2p at 74.1 eV is attributed to γ-Al2O3. The peak at 284.6 eV was attributed to amorphous carbon [47], while the C 1s peaks at about 292–293 eV is a shake-up peak, which is a result of the loss in kinetic energy (increase in binding energy) of the emitted photoelectrons [48]. The analyses of carbon–catalyst samples reveals an additional peak at ca. 288.5–288.7 eV, which is characteristic of carbon atoms involved in inorganic carbonate species or of carbon atoms bonded with several oxygen atoms in carbonate (CO and CO), according to the literature [49–51]. These surface species represent about one third of the carbon present on the surface of the carbon–catalyst samples (Table 1).
The signal corresponding to Pt was very weak due to the relatively low metal content (1 wt.%). In addition, the Al 2p line of the support overlaps the Pt 4f line of the active component, which is usually used for spectroscopic analysis of platinum [52,53]. This overlap makes direct analysis of the platinum states very complicated. Therefore, in this study, we used a different line, Pt 4d. Although this line is weaker, it is not overlapped by the spectral lines of the other components. The presence of the reduced (metal) state Pt°4d5/2 at 314.4–314.7 eV (Table 1) was identified in all the catalysts [54].
The peak located at ca. 529.9–530.5 eV corresponds to O 1s-levels of oxygen atoms O2–in the lattice (Al2O3), but a small contribution could be due to the Pt–O-like species (530 eV) and the Pt–OH ad-species (531.5 eV) [55]. A second peak at ca. 532.7–532.9 eV is observed on the carbon–catalyst sample only. According to the literature [51], we attribute this latter peak to O atoms in the carbonate species rather than to nitrate species as no peak in the N 1s region (400–415 eV) is observed, even on the pure catalyst sample conditioned in the presence of water, for which the x-ray diffractogram revealed the presence of a Ba(NO3)2 phase. Because the XPS analyzes were carried out on samples previously saturated with NOx in the fixed-bed reactor, the absence of N 1s peak, whatever the sample considered, reveals that either nitrate surface species are not stable under ambient atmosphere, leading to the formation of BaO and to a lesser extent barium carbonate species or they were desorbed during the degassing procedure and XPS analysis under low pressure.
The oxide phase (BaO) was detected at a binding energy of ca. 779.7–779.9 eV [52]. The binding energy of Ba 3d5/2 at ca. 780.3–780.4 eV suggests that Ba is present as carbonate. Regarding binding energies of C 1s and Ba 3d5/2 of pure catalyst samples, it is interesting to note too that the presence of water prevents carbonate formation after exposure to ambient atmosphere. In another way, comparison of pure catalyst and carbon–catalyst mixtures conditioned without water, one may observed that barium carbonate species are present in the same extent, while carbonate (CO and CO) is only present on carbon–catalyst mixtures, so that carbonate species formed during the adsorption cycle in the fixed-bed reactor seem not to be in interaction with Ba.
3.3 TEM analysis
TEM images of γ-Al2O3, and the different catalyst and carbon–catalyst samples are presented in Fig. 3. As shown in Fig. 3a, the Al2O3 support consists of a nanofibrous structure a few nanometers thin and tens of nanometers long. TEM images of the catalyst and carbon–catalyst samples show that for these catalysts, several types of crystallites exist: the nanofibers correspond to the γ-Al2O3 support; the round-like crystallites correspond to Pt; and the bulky, large crystallites correspond to Ba carbonate. Among these, Pt forms small spherical particles. There is no homogeneity in particle size; however, the size of Pt is < 3 nm for all catalysts, which is in agreement with the XRD results. The dispersion of these catalysts was approximately 50% TEM images show the region where Ba or C can mask Pt sites and can influence catalytic activity. It is known that small Pt particles (with a high dispersion) are desirable for NO/NOx oxidation, whereas small Pt particles are not desirable for the NSR reaction because they can form aggregates in the presence of oxygen [56,57].

TEM images of γ-Al2O3 (a), pure catalyst sample conditioned in the presence of water in the reactive flow (b), carbon–catalyst sample conditioned in the presence of water in the reactive flow (c), pure catalyst sample conditioned without water in the reactive flow (d) and carbon–catalyst sample conditioned without water in the reactive flow (e).
3.4 DRIFTS experiments
Fig. 4 compares the temporal evolutions of infrared spectra collected during NOx adsorption at 300 °C without water on pure catalyst (a) and carbon–catalyst mixture (b). On pure catalyst (Fig. 4a), as already observed [22], the main process at the beginning is the formation of surface bidentate nitrites (band at 1230 cm−1 with a shoulder at 1320 cm−1), namely (but not exclusively) on Ba sites. After 15–20 min, the nitrite bands decrease in intensity as these species become subsequently oxidized to several kinds of nitrate species:
- • bidentate nMx+ = O2NO– (n = 1,2) (1620–1550; 1290–1240; 1030–990 cm−1);
- • monodentate nMx+ = ONO2– (1520–1460; 1300–1280; 1050 cm−1) on Al and Ba sites;
- • bulk-like (ionic) barium nitrate species (1450–1390; 1360–1300; 1040 cm−1).

(Color online). Time-resolved DRIFTS spectra corresponding to the adsorption of NO (1500 ppm) + O2 (10%) under N2 at 300 °C in the absence (a) and in the presence of carbon black (CB) (b).
When the catalyst is in tight contact with carbon (Fig. 4b), the same evolution of the adsorption band with the time of exposure to NO seems to occur, although the intensity of the band is lower than in the absence of carbon in the sample. The lower intensity (low signal-to-noise ratio) is the consequence of the masking effect of carbon. One may, however, observe the nitrite band (1220–1250 cm−1, as a shoulder) and formation of bidentate nMx+ = O2NO– (n = 1,2) (1620–1550 cm−1) and monodentate nMx+ = ONO2– (1290–1200 cm−1) on alumina and barium as well as bulk-like (ionic) barium nitrate species (1450–1390 cm−1). Subtracting spectra obtained with pure catalyst and with carbon/catalyst mixture after 40 min of exposure under NO gas flow (not shown here) reveals that the carbon/catalyst mixture presents a relatively more important absorption rate than the pure catalyst under the same operating conditions at 1450, 1570 and 1380 cm−1. These adsorption bands are attributed to the formation of carbonates species. These species may be formed either by a direct reaction between nitrite or nitrate species with carbon or by a reaction between nitrite or nitrate species with CO2 formed itself from the oxidation of carbon by O2 and NO2.
3.5 NOx storage capacity
3.5.1 Effect of the presence of CO2 with NO or NO2
Fig. 5 compares the evolution of adsorbed NOx over time during the adsorption phase as a function of the reactive gas’ composition. Although 10% of O2 is injected together with NO, the NOx storage capacity (NSC) after 1 h of exposure to the lean phase increases of about 15% when NO2 is used compared to NO. However, this effect of the reactive gas composition appears only after 800 s of exposure. During the first 800 s of the experiment, the evolution of the amount of stored NOx is similar whatever the nitrogen oxide (NO or NO2) injected. This short period corresponds to the instant the NOx slip reaches 35% of the inlet NOx concentration. According to previous studies focused on the NOx storage mechanism [8,9,11,16], it is generally admitted that NOx adsorption on NSR catalysts occurs with two mechanisms, one characterized by a rapid uptake of NOx leading to nitrite surface species formation, and a second one characterized by a slower rate of NOx uptake leading to the conversion of nitrate into nitrate ad-species. More recently, some authors [8,9] highlighted the participation of two different adsorption sites as a function of their proximity with Pt sites. According to Nova et al. [8], the formation of nitrite species, which are then progressively oxidized into nitrate surface species, occurs on adsorption sites neighboring Pt sites, while on storage sites far from Pt sites, a direct adsorption of NO2 in the form of nitrates takes place. Pt sites play an important role in the oxidation of NO into NO2, as only NO2 is able to be stored on the NSR catalysts. Our results reveal thus that the rapid uptake of NOx occurs on sites close to Pt sites with a similar mechanism as NO or NO2 is injected into the gas phase. In agreement with previous studies [8,16,17,58] and our DRIFT results, this mechanism, reasonably, implies first the formation of nitrite ad-species followed by their oxidation into surface nitrates then bulk species. On the other hand, NOx adsorption on sites far from Pt seems to be favored if NO2 is present in the reactive gas and this although NO is oxidized into NO2 over Pt sites. Thus, using NO as the reactive gas, spillover of NO2 species (derived from NO oxidation on Pt) from Pt to storage sites leads to the formation of stored nitrate species by the same mechanism as when using NO2 as a reactive gas, but migration of NO2 species from Pt to more distant adsorption sites is limited. In our experimental conditions, about 15% of the adsorption sites are thereby not accessible in the presence of NO compared to NO2 in the reactive gas.

Amount of adsorbed NOx versus time over the pure catalyst as a function of the reactive gas composition (300 ppmv NO or NO2, 10% b.v. O2, 0–850 ppmv CO2, 60 nl·h−1).
Injection of 850 ppm CO2 does not influence the amounts and the kinetics of stored NOx. It is moreover important to note that no CO2 emission was detected during the following TPD. This result disagrees with those obtained by Epling et al. [13] or Corbos et al. [1], who observed an inhibiting effect of the presence of CO2 in the gas phase on NSC due to a competition between carbonate and nitrate formation. The lack of inhibiting effect observed in this work is attributed to the low CO2 concentration used (850 ppm vs. 8% to 10% in the other studies). The low CO2 concentration was chosen in this study in order to be representative of CO2 emissions from soot combustion, as one objective of this study is to reveal the influence of the presence of soot in the catalytic bed on the NOx storage mechanism as a function of the reactive gas composition.
NOx emissions profiles, measured during the TPD performed after the adsorption phase, are shown as a function of the temperature in Fig. 6 (solid lines). At low temperature (< 350 °C), NOx emissions are similar whatever the reactive gas composition (NO or NO2) used during the adsorption phase. As is apparent from Fig. 6, at higher temperature, evolutions of NOx are different for the two experiments. TPD performed after an adsorption carried out under NO/O2 shows a nitrate decomposition peak at 410 °C, i.e. 10 °C lower than that obtained after the adsorption was carried out under NO2/O2 as the reactive gas. However, in this latter case, the NOx emission profile is less broad and all nitrates are decomposed at 560 °C, whereas nitrate decomposition is not complete at the end of the heating ramp (600 °C), thus pointing out a more thermal stability of some nitrate species stored in the presence of NO in the reactive gas. In fact, these results suggest that a higher heterogeneity of NOx ad-species is obtained using NO compared to NO2.
NOx outlet during the TPD performed over catalyst (solid line) or carbon–catalyst mixture (dashed line) after NOx adsorption at 300 °C (300 ppmv NO or NO2, 10% b.v. O2).
3.5.2 NOx storage in the presence of carbon black
Table 2 compares the amount of NOx stored on the catalyst with those stored on the catalyst–carbon mixture under the same experimental conditions. The NSC measured when the catalyst is in contact with carbon black (CB) is, whatever the reactive gas composition, lower than those measured in the absence of carbon in the catalytic bed. This result matches those already published [35,40] and confirms that CB inhibits partially the NSC. It was already proposed [35,40] that stored nitrate species on Ba or Al sites, in contact with soot, are destabilized according to the direct surface reactions (1) and (2). This hypothesis is also verified by XRD and XPS characterization, which showed the presence of carbonate species on carbon–catalyst samples. It is interesting to note that the inhibiting effect of CB on NSC (ΔNSC–CB, Table 2) is more important using NO2 than NO in the reactive mixture. Indeed, the decrease of NSC due to the presence of CB reaches 19% using NO and 28% using NO2. Such a result was not observed previously [30]. The major difference between our two works lies in the contact made between CB and the catalyst. In the present study, a tight contact was preferred compared to the loose contact carried out previously [30,54]. Thus, an increase of the carbon–catalyst contact leads to an increase by 50% of the inhibiting effect of carbon on the NSC. The more important NSC loss in the presence of NO2 compared to NO in the reactive gas observed in this work confirms that nitrate species formed on storage sites close to Pt sites suffer to a lesser extent from contact with carbon.
NOx storage capacity (NSC in μmol/gcatalyst) in the presence and in the absence of carbon black under different experimental conditions and rate of loss of NSC due to the presence of carbon (ΔNSC in %) or water (). (300 ppmv NO or NO2, 10% b.v. O2, 0–5% b.v. H2O, 0–850 ppm CO2, 60 nl·h−1).
| Gas composition | NSC (mmol/gcat) | ΔNSC–CB (%) | |||
| Without soot | With soot | Without soot | With soot | ||
| NO2/O2 | 0.567 | 0.410 | 28 | ||
| NO/O2 | 0.476 | 0.384 | 19 | ||
| NO2/O2/H2O | 0.500 | 0.369 | 26 | 12 | 10 |
| NO/O2/H2O | 0.406 | 0.364 | 10 | 15 | 5 |
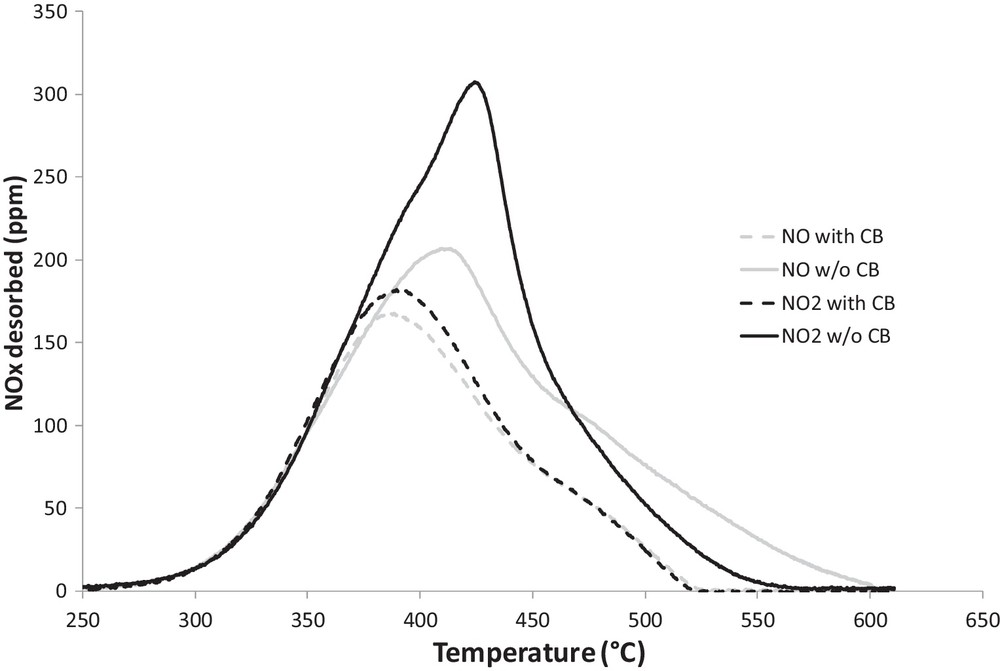
The results of NOx (NO + NO2) emissions during TPD experiments carried out in the absence and in the presence of CB after the adsorption phase performed either with NO or NO2 are compared in Fig. 6. As is apparent from the figure, although the presence of CB shifts the maximum evolution of NOx to a lower temperature reaching thus 385 °C, there is no influence of the presence of CB on the amount of NOx desorbed at low temperature (< 350 °C). The temperature threshold for nitrate decomposition equals 250 °C, so below the NOx adsorption temperature (300 °C), in the absence as in the presence of carbon in the catalytic bed. Although NOx desorption is not affected by the presence of carbon at low temperature, a modification of the repartition of the decomposition products between NO and NO2 is observed in the presence of CB and this on the whole temperature range (250 to 600 °C). Fig. 7 (compare grey solid and dashed lines) shows that CB enhances significantly NO (instead of NO2) release. In fact, in the presence of CB, NO emissions are shifted to lower temperature on the whole temperature range (250 to 600 °C), whereas NO2 releases begin at higher temperatures. This phenomenon is particularly important when the adsorption phase occurred under an NO2/O2 mixture. Actually, NO2 threshold temperature is shifted to higher temperature when the catalyst is in contact with carbon, but the maximum emission of NO2 is shifted from 15 °C to a lower temperature and no NO2 emission is observed from 450 °C onward, whereas in the absence of carbon in the catalytic bed some NO2 emissions are measured until 500 °C. In order to understand if the presence of carbon influences the initial decomposition of nitrate species and in what extent decomposition of nitrate species allows carbon oxidation, NO and CO2 emissions measured during the beginning of the TPD (T < 350 °C), performed with the catalyst–carbon mixture, were compared (Fig. 8). One may observe that CO2 emissions are, in this temperature range (200 °C–350 °C), in correspondence with NO emissions. Indeed, until 340 °C when the adsorption phase is performed using NO, and 350 °C using NO2, NO emissions are twice those of CO2. This result, together with the absence of influence of CB observed on the overall amount of NOx emission during TPD in the temperature range 200–340 °C, leads us to conclude that, in this temperature range, CB has no direct influence on nitrate decomposition. Conversely, NO2 emitted, in the gas phase, from thermal nitrate decomposition allows carbon oxidation according the well-known reaction:
| (3) |

NO (a,c) and NO2 (b,d) outlet during the TPD performed over the catalyst (solid lines) or the carbon–catalyst mixture (dashed lines) after NO (a,b) ou NO2 (c,d) adsorption at 300 °C (300 ppmv NO or NO2, 10% b.v. O2, 0 or 5% b.v. H2O).

NO and CO2 emissions during the TPD at low temperature. TPD was performed after NOx adsorption at 300 °C (300 ppmv NO or NO2, 10% b.v. O2).
At higher temperature (> 350 °C) the shapes of NO and NO2 releases are almost similar in the absence and in the presence of carbon, but shifted to lower temperature when carbon is present in the catalytic bed. CO2 emissions present a maximum at 380 °C, then stabilize at a constant value of about 40 ppm between 450 °C and 520 °C, and finally progressively increase until the end of the heating ramp (600 °C) before decreasing (not shown here). In Fig. 9 is represented the difference between NOx emissions measured during the TPD performed, after adsorption under NO/O2 as reactive gas, on the pure catalyst and carbon–catalyst samples (solid black line). The obtained curve presents three peaks: a negative one in the 340–375 °C temperature range, with a maximum at 365 °C, and two positive ones at 420 °C and 520 °C. The negative peak points out an enhancement of nitrate decomposition by CB. Since the latter is accompanied by an increase of CO2 emissions (Fig. 8), it seems that an interaction between CB and adsorbed nitrates occurs, leading to both decomposition of nitrate and oxidation of carbon. Matarrese et al. [33,59], who studied the decomposition of nitrate species on K- or Ba-based NSR catalysts, already highlighted, as what is observed in this study, that the presence of soot in contact with a NSR catalyst leads to a modification in the distribution of products from the thermal decomposition of adsorbed nitrate species. In their experimental conditions, these authors also observed, in contrast to our study, that decomposition of nitrates at low temperature is shifted at lower temperatures (< 350 °C). They attributed these observations to a direct surface reaction between stored nitrates and soot without the necessity of preliminary thermal decomposition of nitrates species. As this effect is more important in the case of K-based catalysts than in the case of Ba-based catalysts, the surface reactions proposed are different between K-based and Ba-based catalysts. In the first case, the surface reaction would favor carbon oxidation, while using the Ba-based catalyst, the nitrate species would react with soot, leading to carbonates species. According to Matarese et al. [33,59], it seems that in our study, a surface reaction between CB and stored nitrates occurs. However, this enhancement of nitrate decomposition by carbon is limited in the 340–375 °C temperature range, and, contrary to what was proposed by Matarese et al. [32,41] using a Ba-based catalyst, this interaction enhances both nitrate decomposition and carbon oxidation. Hence the following reaction is proposed:
| (4) |

Difference between NOx emissions measured during the TPD performed on the catalyst alone or mixed with carbon after NO adsorption at 300 °C (300 ppmv NO, 10% b.v. O2).
It is important to note that the beneficial interaction between carbon and catalyst in the decomposition of nitrate species, according to (4), affects only a small proportion of the adsorbed NOx. Indeed, from Fig. 9, the amount of stored NOx whose decomposition is promoted by the presence of carbon represents only 4% of the adsorbed NOx.
The second peak observed at 420 °C (Fig. 9) is attributed to the decomposition, of the nitrate species that had not been formed during the lean phase when this one is performed in presence of carbon in the catalytic bed. This decrease of nitrate formation during the adsorption phase is the consequence of the carbon–catalyst interaction and thus carbonate formation according to (1) and (2) as observed in Table 2. According to the literature [17,60–62], given the temperature range where these nitrate species decompose, it seems that these species are surface nitrate species. One may then conclude that nitrate species that are destabilized by carbon at 300 °C during the adsorption phase are predominantly surface nitrate species. Finally, the third peak, observed at 520 °C, is attributed, in agreement with previous work [40], to both the complement part (∼30%) of the inhibition of NOx storage on Ba sites during the adsorption phase due to the presence of carbon and to a reduction of the NOx stored into the Ba(NO3)2 bulk formed during their desorption according to the following equation:
| (5) |
Indeed, it should be noted here that contrary to what observed in the absence of carbon black in the reactor, the nitrogen balance from the quantity of NOx adsorbed during the lean phase and NOx desorbed during TPD was not obtained for experiments performed in the presence of CB. Hence, the amount of NOx (NO + NO2) desorbed during the TPD corresponds to 80% of the amount stored during the adsorption phase. Therefore, in the presence of carbon, a non-negligible amount of stored NOx was reduced during the TPD. On Fig. 6 it is able to observe that this reduction occurs at high temperature (no NOx emission occurs at temperature higher than 530 °C when carbon is present in the catalytic bed).
3.5.3 Influence of H2O
The influence on the NSC of the injection of 5% H2O together with 300 ppm NO and 10%O2 during the adsorption phase performed either in the absence or in the presence of carbon in the catalytic bed is shown in Table 2.
In the absence of CB, a decrease of 12% to 15% of the NSC is observed when water is present in the reactive gas. Moreover, a decrease of the NO2/NOx ratio, measured at the end of the adsorption phase performed under NO/O2, from 0.59, when no water is injected, to 0.41, in the presence of water, was observed.
Fig. 7 compares (solid lines) NO and NO2 releases during the TPD performed after the adsorption phase, with NO (Fig. 7a and b) or NO2 (Fig. 7c and d) in the absence or the presence of water in the reactive gas. When water is present in the lean phase, the threshold temperature, as well as the maximum, of NO2 release is shifted to higher temperatures (about 10 °C). In the same way, a significant increase in the NO emissions at high temperatures (> 400 °C) is observed when the TPD is performed after an adsorption phase under a gas flow containing water. Since XRD characterization showed the formation of crystals of barium nitrate on pure catalyst or carbon–catalyst mixture only after they are conditioned under a water-containing gas flow, and in accordance with our previous work [22], it can be concluded that NOx adsorption in the presence of water leads, of course, to a decrease in the NSC, but also promotes the formation of more stable nitrate species, i.e. bulk species, at the expense of surface nitrates species. These results agree with the well-known partial inhibiting effect of water on the NSC of NSR catalysts at low temperatures (< 300–350 °C) [1,12,13,15,20]. The observed decrease of the NO2/NOx ratio during the adsorption phase is in agreement with works [12,15,20,22] showing that water affects particularly the oxidation of NO into NO2 over Pt sites. The decrease in the NSC observed in the presence of H2O in the gas flow is thus firstly attributed to a decrease of the local NO2 concentration. It was moreover established that H2O inhibits particularly the formation of nitrites species on Ba sites and so the formation of surface nitrate species [16,17,22] while, on the other hand, the formation of bulk nitrate species was proposed to be enhanced when water is present in the reactive gas [22,61].
In the presence of CB in the catalytic bed, Table 2 reveals that the presence of water in the reactive gas during the adsorption phase decreases the inhibiting effect of CB on NOx adsorption. Indeed, the loss of NOx stored in the presence of carbon decreases from 19% to 10% and 28% to 26% when the adsorption is performed under NO and NO2, respectively. In the same way, when carbon is in contact with the catalyst, the inhibiting effect of water on the NSC decreases too: from 15% to 5% and 12% to 10% as the adsorption is performed under NO or NO2, respectively. Fig. 7 confirms the opposite effect of the presence of CB and water on the NO and NO2 emissions during the TPD. Based on the results presented above, one may propose that the non-cumulative effect of CB and water is the result of a competition between the destabilization of weakly bond surface nitrate species by CB according to (1) and (2) and the enhancement of bulk nitrate species formation from weakly bonded surface nitrate species by water. The simultaneous effect of water and CB is summarized in Scheme 1.

Competitive effect of CB and water.
4 Conclusion
In this work, the influence of the presence of carbon in the catalytic bed and/or water in the gas phase on the NOx storage capacity of a model four-way catalyst (1%Pt–10%Ba/Al2O3) was investigated. Based on XRD, XPS and DRIFTS characterizations, it was confirmed that, when carbon is present in the catalytic bed, the decrease in NSC is the consequence of carbonate formation according to a direct surface reaction between carbon particles and stored nitrate species. A careful analysis of NO and NO2 release during TPD revealed that only surface nitrate species that decomposed at about 420 °C are involved in this process. Moreover, from NO/O2 and NO2/O2 adsorption experiments, it was observed that nitrate species formed on storage sites close to Pt suffer to a lesser extent from contact with carbon.
From the TPD analysis, it was shown that the presence of CB in the catalytic bed does not influence the decomposition of very weakly bonded nitrates or nitrites species that decompose at temperature lower than 340 °C. At higher temperature, the decomposition of a small part of the stored NOx is improved by carbon, which is itself oxidized according to:
Adsorption tests showed that the presence of H2O in the gas flow not only decreases NSC, but also promotes the formation of more stable nitrate species. XRD characterization corroborates this result, as a crystalline barium nitrate phase was observed on samples conditioned with water instead of an amorphous form when adsorption is performed without water.
Finally, a non-cumulative effect of carbon and water was observed. It was proposed that a competition between the destabilization, by carbon, of weakly bonded surface nitrate species and the enhancement of bulk nitrate species formation in the presence of water occurs.
Acknowledgments
The authors thank the network REALISE (Réseau Alsace de Laboratoires en Ingénierie et Sciences pour l’Environnement) for its financial support for equipment facilities. Financial support by the CNRS France is gratefully acknowledged by Ioana Fechete. The CSC (China Scholarship Council) is acknowledged by Dongliang Wu for its financial support.