

1 Introduction
Organometallic complexes involving the dianionic Cot ligand (Cot = η8-C8H8) are predominant species in the f-element chemistry since Streitwieser et al. reported the uranium compound [U(Cot)2], the first analogue of ferrocene with a C8 aromatic ligand, in 1968 [1,2]. The discovery of this linear and highly symmetric sandwich compound, rapidly followed by the synthesis of the lanthanide (Ln) and actinide (An) derivatives [Mf(Cot)2]q− (Mf = Th, Pa, Np, Pu, Ce and q = 0 [3–5]; Mf = Ln, U, Th, Pu and q = 1 [2,6,7]), was an experimental and theoretical entry for a better understanding of the Mf− ligand bonding. The weak reactivity of the uranium compound, in particular, induced considerable debates and interest [3,8]. For 40 years, the reluctance of the [Mf(Cot)2]q− (q = 0, 1) species to ligand coordination, attributed to the steric protection of the two parallel Cot ligands, clearly evidenced the poor reactivity of these compounds and their inability to form bent derivatives [9].
However, the first bent [Mf(Cot)2] complexes, obtained through two different strategies, were recently characterized. In 2008, uranocene was found to capture the cyanide ion, a strongly coordinating ligand with appropriate small size, to give the mono cyanide derivative [U(Cot)2(CN)][NR4] [10]. In 2011, the free adducts [Mf(1,4-{Ph3Si}2C8H6)2]q− (Mf = Ce and q = 1, Mf = U and q = 0), were also found to adopt a bent geometry as the result of steric constraints imposed by the very bulky Ph3Si substituents in 1,4 positions of the Cot ring [11]. These results incited us to re-examine the coordination chemistry of tetravalent actinocenes (U, Th) towards neutral and anionic ligands.
Because [U(Cot)2] proved too stable and unreactive with ligands less coordinating than cyanide, we replaced U4+ by the larger Th4+ ion (r(Th4+) ∼ r(U4+) + 0.05 Å) [12], and focused our interest on [Th(Cot)2] (1) which has been far less studied than its uranium counterpart. Some chemical features however emerged, which differentiate thorocene from its uranium congener [13–15], such as the more facile hydrolysis, the ready formation of the mono-Cot compound and the lower thermodynamic stability as observed through competition reactions of the substituted thorocene and [UCl4]. These differences, attributed to the less covalent Th − Cot bonding [15,16], led us to suppose that further distinct chemical behavior could be revealed. By considering the more ionic character in thorocene, a greater mobility of the Cot ligand around the metal centre was expected, that should be more favorable to ligand coordination.
In that perspective, a series of coordinating molecules have recently been reacted with thorocene, such as isonitrile and aromatic nitrogen molecules [17] and proved to be efficient wedges to open this molecular sandwich, leading to unique neutral [Th(Cot)2(L)] complexes with bent geometry (Scheme 1). In the presence of phenanthroline (phen) or its tetramethyl derivative, the much more stable compounds [Th(Cot)2(R4phen)] (R = H, Me), were isolated and the crystal structure clearly revealed the bidentate coordination mode of the ligand, thus, demonstrating that not only one but two coordination sites are available on the bent thorocene fragment (Scheme 1).
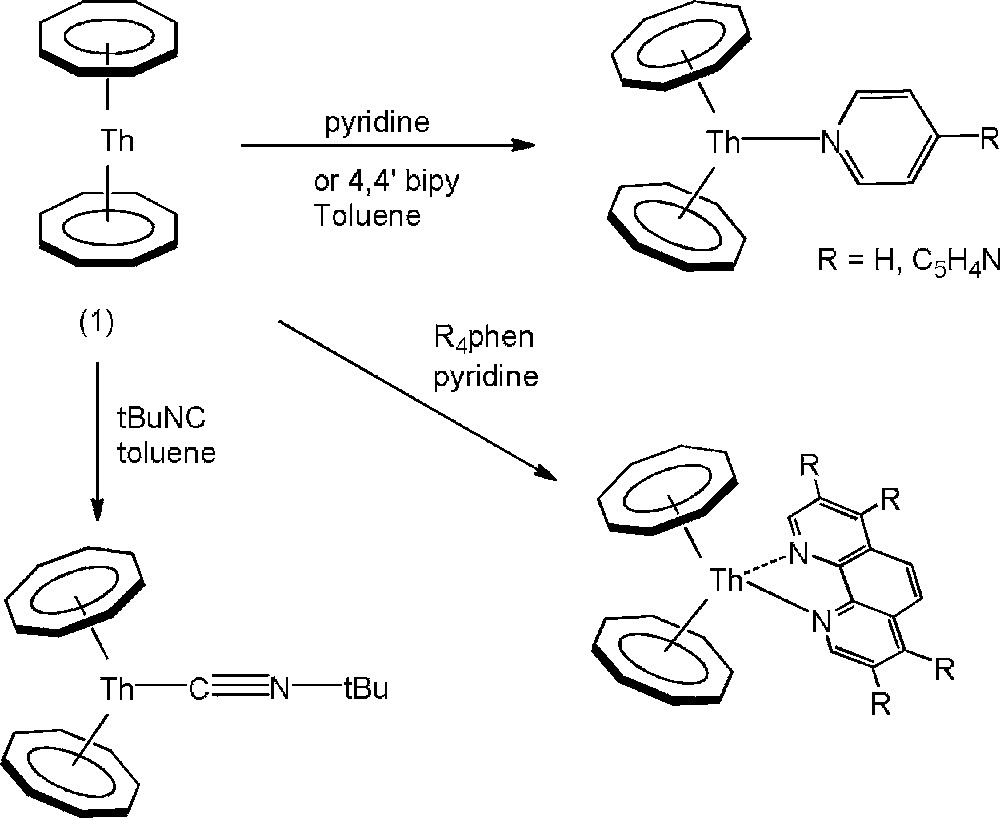
Reactions of 1 with neutral Lewis bases.
Nitrogen molecules are ubiquitous ligands in coordination chemistry. They can modulate the electronic richness of the metal centre through their donor and π-accepting properties and provide interesting redox activity. They are also of importance in various fields from extraction processes to the elaboration of materials with specific luminescent and/or magnetic properties. However, it is noteworthy that in contrast to that observed with d-transition metals, 2,2′-bipyridine complexes of the actinides are not common [18–20], particularly, those involving the reduced (bipy)− or (bipy)2− ligand [21].
Here, we report that, in striking difference with uranocene, bending of the [Th(Cot)2] fragment is readily achieved with 2,2′-bipyridine (bipy) to give [Th(Cot)2(κ2-bipy)], which can be easily reduced into [Th(Cot)2(κ2-bipy)]−, a thorium(IV) complex involving the bipy− radical anion.
2 Experimental
2.1 Materials and reagents
The complexes described below are moisture sensitive. Syntheses and manipulations of the compounds were conducted under ultra-high purity argon atmosphere with rigorous exclusion of air and water, using Schlenk-vessel and vacuum-line techniques or in a glove box. The solvents, toluene, pentane and tetrahydrofuran, were dried over a mixture of sodium-benzophenone and pyridine over potassium, and were distilled immediately before use. Deuterated pyridine (Eurisotop) was distilled over K and stored over 3 Å molecular sieves in the glove box. K2Cot [2,22], [ThCl4(dme)2] [23] and [U(Cot)2] [1,2] were prepared as previously described and [Th(Cot)2] (1) [13] was obtained from a modified method starting from [ThCl4(dme)2][17]. 2,2′-Bipyridine (bipy), was purchased from Aldrich, dried under vacuum and stored under argon. The 1H NMR signal of K2C8H8 is at δ 6.67 in [D5]pyridine and δ 5.67 in [D8]THF and that of C8H8 is at δ 5.64 in [D5]pyridine [24]. The 1H NMR signals of pyridine were referenced at δ 8.57, 7.41 and 7.0. IR samples were prepared as Nujol mulls between KBr round cell windows and the spectra recorded on a PerkinElmer FT-IR 1725X spectrometer. The 1H and 13C{1H} NMR spectra were recorded at 23 °C on a Bruker DPX 200 instrument and referenced internally using the residual protio solvent resonances relative to tetramethylsilane (δ 0). Elemental analyses were performed by Analytische Laboratorien at Lindlar (Germany) or by Medac Ltd at Chobham (Surrey, UK). The systematic low carbon content in the analytical results for compounds 2 and 3 is presumably due to thorium carbide formation [15,17,25].
Caution! Depleted uranium (primary isotope 238U) is a weak α-emitter (4.197 MeV) with a half-life of 4.47 × 109 years; natural thorium (primary isotope 232Th) is a weak α-emitter (4.012 MeV) with a half-life of 1.41 × 1010 years; all the manipulations and reactions should be carried out in monitored fume hoods or in a glove-box in a laboratory equipped with α- and β-counting equipment.
2.2 Syntheses
2.2.1 Synthesis of [Th(Cot)2(bipy)] (2)
A 25-mL round-bottom flask was charged with 1 (150 mg, 0.34 mmol), bipy (54 mg, 0.34 mmol) and acetonitrile (5 mL). A brown solid was immediately formed. After 1 h at 60 °C, this solid was filtered off, washed with toluene (2 × 5 mL) and dried under vacuum for 5 h at 23 °C (198 mg, 97%). C26H24N2Th requires: C 52.35, H 4.06, N 4.70; found C 51.23, H 4.18, N 5.05. 1H NMR ([D5]pyridine, 23 °C): δ = 8.61 (broad, 2 H), 8.28 (d, J = 4 Hz, 2 H), 7.78 (t, J =7 Hz, 2 H); 7.33 (signal partially masked by py, 2 H), 6.14 (s, 16 H, Cot);13C{1H} NMR: δ = 148.43, 138.98, 125.72, 122.51, 102.8(Cot). IR (cm−1): 1863 m, 1747 m, 1602 s, 1561 w,1315 s, 1178 m, 1157 s, 1100 w, 1011 s, 922 s, 784 m, 790 s, 777 s, 725 s, 695 s, 642 w. The IR spectrum of [Th(Cot)2] is: 1865s, 1750s,1631 m, 1602 m, 1316s, 1178w, 1082w, 897s, 791 m, 778 m, 743s, 697s, 507w. Black crystals of 2 were obtained the by slow diffusion of Et2O into a pyridine solution, or by heating a MeCN or THF suspension of 2 at 90 °C for 15 h.
2.2.2 Synthesis of [Th(Cot)2(bipy)][K] (3)
A 25-mL round-bottom flask was charged with 2 (80 mg, 0.13 mmol), KC8 (25 mg, 0.18 mmol) and THF (5 mL) immediately affording a dark-red solution with a black C8 deposit. After 1 h at 23 °C, the solution was filtered and the solvent evaporated off, giving 3 as a dark-brown solid (81 mg, 95%). C26H24N2ThK requires: C 49.13, H 3.81, N 4.41; found: C 47.97, H 4.21, N 4.37. The 1H NMR spectra at 23 °C in [D8]THF or [D5]pyridine do not show any signal for Cot and bipy ligands. 13C{1H} NMR ([D8]THF, 23 °C): 91.45, 66.13, 16.04. IR (cm−1): 1865 m, 1815w, 1743 m, 1677 m, 1582s, 1542s, 1491s, 1279s, 1260s, 1212s, 1143s, 1106s, 1082s, 1046 m, 1014s, 1003s, 949s, 900s, 836s, 784 m, 749s, 706s, 677s, 641s, 603 m, 551 m, 446 m, 424 m. Black crystals of 3(py)2 were obtained by slow diffusion of Et2O into a brown pyridine solution of 3.
An NMR tube was charged with bipy (4.0 mg, 0.025 mmol), KC8 (3.5 mg, 0.025 mmol) and [D8]THF (0.5 mL). The solution immediately turned purple and the 1H NMR spectrum did not show the signals for reduced bipy. Addition of yellow 1 (11.2 mg, 0.025 mmol) induced instantaneous formation of a dark brown precipitate. No 1H NMR signal, except for a weak signal related to free C8H82− at δ 5.68.
2.2.3 Reaction of 3 with TlBPh4
An NMR tube was charged with 3 (11.5 mg, 0.018 mmol), TlBPh4 (9.5 mg, 0.018 mmol) in [D5]pyridine (0.5 mL). Immediately, a grey powder of Tl(0) was observed. The 1H NMR spectrum showed the signals relative to 2.
2.2.4 Treatment of [U(Cot)2] with bipy
An NMR tube containing [U(Cot)2] (10 mg, 0.022 mmol), bipy (3.5 mg, 0.022 mmol) and [D8]THF (0.5 mL) was heated at 90 °C. After 1 h, the pale green solution contained a large quantity of poorly soluble [U(Cot)2] and the 1H NMR spectrum revealed the presence of free bipy and [U(Cot)2]. After addition of 1 equiv KC8 (3.4 mg), the solution turned purple (Kbipy) and then became darker. After 1 h at 90 °C, the 1H NMR spectrum only showed the presence of K2Cot (δ = 5.68), free bipy, [U(Cot)2] and other unidentified paramagnetic Cot signals. The presence of a green powder of uranocene was observed.
2.3 Crystallography
The data were collected at 150(2) K on a Nonius Kappa-CCD area detector diffractometer [26] using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The crystals were introduced into glass capillaries with a protecting “Paratone-N” oil (Hampton Research) coating. The unit cell parameters were determined from ten frames, then refined on all data. The data (combinations of φ- and ω-scans giving complete data sets up to θ = 30°C and a minimum redundancy of 4 for 90% of the reflections) were processed with HKL2000 [27]. Absorption effects were corrected with SCALEPACK [27]. The structures were solved by direct methods with SHELXS-97, expanded by subsequent Fourier-difference synthesis and refined by full-matrix least-squares on F2 with SHELXL-97 [28]. All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were introduced at calculated positions and were treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom. The drawings were done with ORTEP-3 [29]. Crystal data and structure refinement parameters are given in Table 1 and the selected bond lengths and angles are listed in Table 2. CCDC-920364 and 920365 contain the supplementary crystallographic data for compounds 2 and 3(py)2. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data and structure refinement details.
| 2 | 3(py)2 | |
| Chemical formula | C26H24N2Th | C36H34KN4Th |
| M (g mol−1) | 596.51 | 793.81 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/m |
| a(Å) | 8.6148(2) | 10.4981(5) |
| b(Å) | 31.8694(4) | 9.3769(2) |
| c(Å) | 14.8246(3) | 15.0666(7) |
| β(°) | 93.7902(5) | 98.436(2) |
| V(Å3) | 4061.17(14) | 1467.10(10) |
| Z | 8 | 2 |
| Dcalcd (g cm−3) | 1.951 | 1.797 |
| μ(Mo Kα) (mm−1) | 7.358 | 5.257 |
| F(000) | 2272 | 774 |
| Reflections collected | 93454 | 54986 |
| Independent reflections | 12383 | 4721 |
| Observed reflections [I > 2σ(I)] | 10609 | 4199 |
| Rint | 0.039 | 0.068 |
| Parameters refined | 523 | 218 |
| R1 | 0.025 | 0.024 |
| wR2 (all data) | 0.055 | 0.044 |
| S | 0.988 | 0.973 |
| Δρmin (e Å−3) | −2.14 | −1.47 |
| Δρmax (e Å−3) | 1.10 | 0.76 |
Selected bond lengths (Ǻ) and angles (°) in 2 and 3(py)2.
| 2 | 3(py)2 | |
| Th–C | Th1–C11 2.787(3); Th1–C12 2.784(3); Th1–C13 2.774(3) | Th–C11 2.828(3) |
| Th1–C14 2.822(3); Th1–C15 2.865(3); Th1–C16 2.866(3) | Th–C12 2.816(3) | |
| Th1–C17 2.813(2); Th1–C18 2.788(2); Th1–C19 2.794(3) | Th–C13 2.889(2) | |
| Th1–C20 2.788(2); Th1–C21 2.784(2); Th1–C22 2.833(3) | Th–C14 2.945(2) | |
| Th1–C23 2.892(3); Th1–C24 2.860(3); Th1–C25 2.805(3) | Th–C15 2.913(3) | |
| Th1–C26 2.796(3) | Th–C16 2.847(3) | |
| Th2–C37 2.789(3); Th2–C38 2.804(3); Th2–C39 2.795(3) | Th–C17 2.840(3) | |
| Th2–C40 2.816(3); Th2–C41 2.866(3); Th2–C42 2.856(3) | Th–C18 2.833(3) | |
| Th2–C43 2.806(3); Th2–C44 2.784(3); Th2–C45 2.799(3) | ||
| Th2–C46 2.773(2); Th2–C47 2.767(3); Th2–C48 2.828(2) | ||
| Th2–C49 2.898(2); Th2–C50 2.892(3); Th2–C51 2.814(2) | ||
| <Th–C(Cot)> | 2.82(4) | 2.86(4) |
| Th···Cga | 2.14/2.15 | 2.20 |
| Th−N | Th1−N1 2.732(2); Th1−N2 2.700(2) | 2.549(2) |
| Th2−N3 2.693(2); Th2−N4 2.687(2) | ||
| <Th–N> | 2.703(17) | |
| N1–Th–N2 | 59.11(6)/59.95(6) | 62.75(7) |
| Cg1···Th···Cg2 | 143.7/142.9 | 140.0 |
| Cg···K···Cg | 131.6 | |
| K−N, K−C | K–N3 3.009(3) | |
| K–N4 3.184(3) | ||
| K–C11 3.414(3) | ||
| K–C12 3.338(3) | ||
| K–C13 3.212(3) | ||
| K–C14 3.176(3) | ||
| K–C15 3.368(3) |
a Cg is the centroid of the cyclooctatetraenyl ring.
3 Results and discussion
3.1 Synthesis and characterization
In contrast to uranocene, which we found inert in the presence of bipyridine, [Th(Cot)2], (1) [13] reacted immediately with 1 equiv bipy in THF or acetonitrile to give immediately a brown powder which was filtered off and washed with toluene. The 1H NMR spectrum in pyridine, which shows a diamagnetic Th–Cot signal at δ 6.14, shifted upfield in comparison to that of 1 (δ 6.28) and new signals corresponding to bipy, suggests the formation of [Th(Cot)2(k2-bipy)] (2). Black crystals suitable for X-ray diffraction were obtained by heating a suspension of the product in THF or acetonitrile or by slow diffusion of Et2O into a pyridine solution of 2 (Scheme 2).
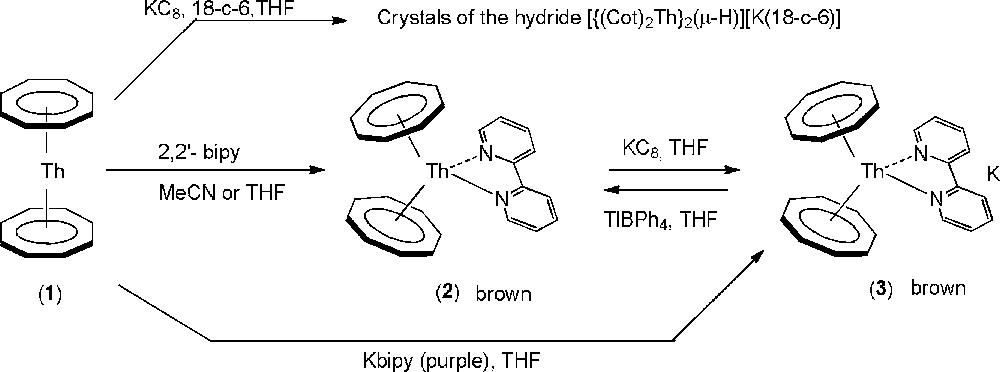
Synthesis of 2 and 3.
The crystal structure of 2 clearly evidences the coordination of one bipy in a bidentate fashion onto an unusual bent “actinocene” [An(Cot)2] fragment [17,30]. After [Th(Cot)2(Me4Phen)] [17], 2 is the second example of an [An(Cot)2(L)2] species. Under similar conditions, [Th(Cot)2] only retains a single pyridine molecule whereas there is space enough for two [17], which shows the importance of chelation so as to get a larger coordination number in complex 2. After [(Cp′′)2Th(F)(μ-F)2Th(Cp′′)(F)(bipy)] (Cp′′ = η5-1,3-(Me3C)2C5H3), 2 is the second structurally characterized Th-bipy compound [21b].
A special attention has been recently paid to the synthesis and physicochemical properties of unusual low-valent lanthanide and actinide compounds or their synthetic equivalents containing reduced ligands [31], such as aromatic polydentate nitrogen ligands [18a,18g,32]. Anionic bipy•− or bipy2− are such redox active ligands favouring reduction chemistry [21]. It was then of interest to consider the reduction of 2 into the formally Th(III) derivative [Th(Cot)2(bipy)]−, that would open attractive chemical perspectives and further comparisons with the corresponding bent metallocene species [(Th(C5H5-nRn)2(bipy)] involving the bipy2− ligand [21a–c].
In contrast to [Th(C8H6{SitBuMe2}2)2][K(DME)2], which is a stable green Cot-substituted derivative of the trivalent thorocene [6], we could not get [Th(Cot)2]−, the treatment of 1 with K(18-crown-6)C8 in THF leading only to the reproducible formation of pale green platelets of the bimetallic hydride [{Th(Cot)2}2(μ-H)]–[30]. This latter could result from the transient formation of [Th(Cot)2]−, which would be much less stable than [Th(C8H6{SitBuMe2}2)2][K(DME)2] [6], which is stabilized by the steric and electron attracting effects of the SitBuMe2 substituents. In contrast, 2 was found to react rapidly with KC8 in THF to give immediately a dark-red solution of the expected [Th(Cot)2(bipy)][K] (3), with the precipitation of graphite. Usual work-up afforded a dark-brown solid, which is 1H NMR silent but whose analysis is in agreement with the formula of 3. The reaction is reversible and the oxidation of 3 with TlBPh4 gave back 2 almost quantitatively as observed by 1H NMR. The infrared spectrum of 3 suggests the formation of a Th(IV)-(bipy−) species rather than a true Th(III) complex with neutral bipyridine since it contains a strong band at 949 cm−1 and vibrations at 1542 and 1582 cm−1 consistent with a reduced bipyridyl radical anion [21d]. Neutral bipyridine is known to display a strong absorption at ∼1600 cm−1 without strong bands in the 900–1000 cm−1 range, whereas the bipyridyl radical anion shows an absorption near 900–1000 cm−1 due to the ring deformation of the ligand, and several CC and CN stretching frequencies in the 1475–1625 cm−1 region [21d]. At last, the formation of 3 was definitively evidenced by growing black crystals of {[Th(Cot)2(bipy)K(py)2]}∞ (3(py)2) by the slow diffusion of Et2O into a pyridine solution of 3.
3.2 Crystal structures of [Th(Cot)2(bipy)] (2) and {[Th(Cot)2(bipy)K(py)2]}∞ (3(py)2)
Views of the structures of 2 and 3 are shown in Figs. 1 and 2, respectively. Both 2 and 3 contain a bent thorocene fragment coordinated with a bidentate and almost planar (bipy)q− ligand. The two pyridyl moieties of the bipy ligand in 2 form a small dihedral angle of 3.9(2)° but they are perfectly co-planar in 3. If the neutral compound 2 is monomeric, the anionic derivative 3(py)2 is a polymer due to the bridging role of the K(py)2+ group. As represented in Fig. 2b, 3 forms an infinite chain where the [Th(Cot)2(bipy)]− anions are linked by cationic K(py)2+ groups sandwiched in η5-fashion by two Cot ligands from two adjacent [Th(Cot)2(bipy)] anions. Mixed sandwich complexes involving M(Cot)Ln(Cot) linkages are uncommon (M = Li, K, Na) [33] and 3(py)2 is the unique f-element species containing a bent (C8H8)2K moiety whereas this latter is linear in the tetrametallic complex [(Cot)Er(Cot)K(Cot)Er(Cot)K(THF)4] [33a]. Five K–C distances are in the range 3.212(2)–3.414(3) Å while the other three distances are somewhat larger at 3.525(3)–3.596(3) Å; these values are larger than those of 2.985(10)–3.018(25) Å in [{K(THF)3(μ-η8:η8-Cot)}2Eu] [33d] and 2.936(6)–3.029(7) Å in [K2(THF)3(μ-η8:η8-Cot)] [34]. Along the chain, the pyridine and the bipy ligands are in opposite positions and the (Th, N1, N2) and (K, N3, N4) groups of atoms form parallel planes (Figs. 1 and 2).
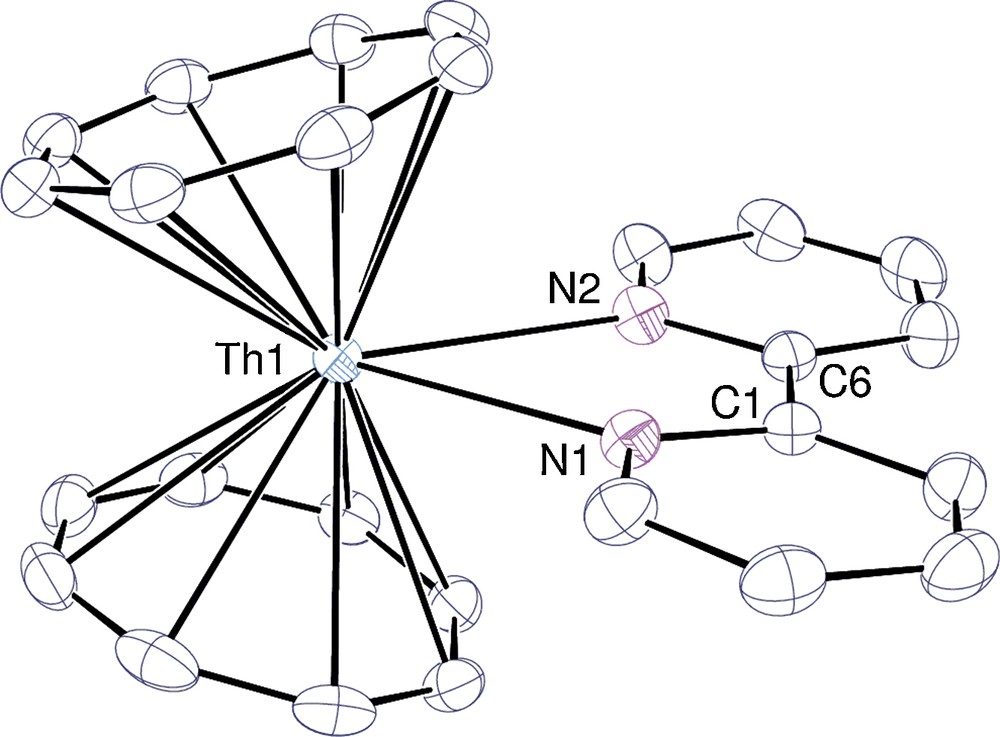
View of one of the two independent molecules in [Th(Cot)2(κ2-bipy)] (2) with displacement ellipsoids drawn at the 40% probability level and hydrogen atoms omitted.

(a) View of [Th(μ-η8:η5-C8H8)2(κ2-bipy)K(py)2]∞ (3(py)2) with displacement ellipsoids drawn at the 40% probability level and hydrogen atoms omitted. (b) View of the 1D polymer (green: K+, blue: Th4+).
The carbon atoms of the C8 rings of 2 and 3(py)2 are co-planar with a maximum rms deviation of 0.037 Å and the Cg···Th···Cg angle (Cg = ring centroid) deviates notably from linearity, with values of 143.7 [142.9] and 140.0°, respectively. These angles are quite smaller than those found in the complexes [Th(Cot)2(L)]q− with monodentate L ligands (L = tBuNC, py, 4,4’-bipy, CN−, N3−) which are in the range [148°–154.3°] or in the bimetallic [{Th(Cot)2}2(μ-H)][Na(18-crown-6)(THF)2] (148°) [17,30]. The angle in 2 is comparable to that measured in [Th(Cot)2(Me4phen)] (144.2°), whereas the smaller value in 3(py)2 can be related to an excess of charge within the bipy ligand, leading to a greater electronic repulsion with the Cot groups. These angles are notably larger than those found in the metallocenes [Th(C5H2tBu3)2(bipy)] [134.5(5)°] and [Th(C5H3tBu2)2(bipy)] [124.1(8)°] containing the bipy2− dianion [21a–c]. In comparison and as a result of the larger ionic radius of the potassium ion, the Cg···K···Cg angle in 3(py)2 is smaller, with a value of 131.6° (Fig. 3).
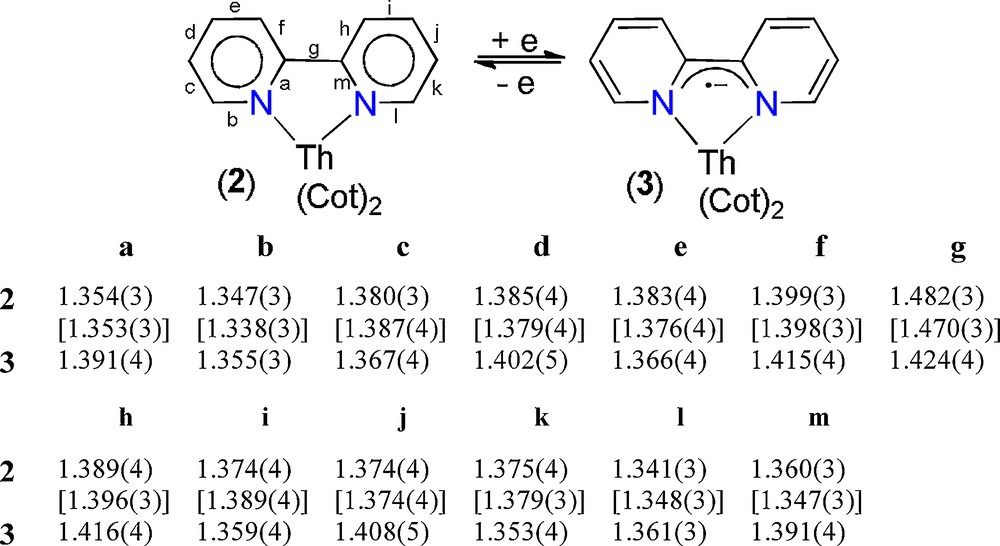
Bond lengths (Å) within the bipy and (bipy)•− ligands in 2 and 3(py)2.
The average Th–C(Cot) distance of 2.82(4) Å in 2 (the same in the two independent molecules) is close to that in 3(py)2 [2.86(4) Å] and larger than that in 1 [2.701(10) Å] [14]. However, the range spanned in the ten-coordinate linear parent 1 [2.679(14)–2.714(12) Å] is narrower than in 2 [2.767(3)–2.898(2) Å] and 3(py)2 [2.816(3)–2.945(2) Å]. The larger Th–C(Cot) distances in 3(py)2 by comparison with those in 1 and 2 reflect the bridging vs the terminal position of the Cot ligands. The mean Th–C(Cot) distance in 3(py)2 can also be compared with that of 2.789(6) Å (for the capped Cot ligand) in the ten-coordinate trivalent complex [(C8H6{tBuMe2Si}2)2Th)[K(DME)2] [6]. These differences also reflect the variation in the coordination numbers and the charge of the complexes.
The geometrical parameters of the bidentate bipy ligand (C–C/N distances listed in Fig. 3 and dihedral angles) coupled with the M–N bond lengths are generally good indicators to differentiate neutral or reduced bipyridine. The X-ray data on 3(py)2 support the infrared data in showing that a reduced bipyridyl radical anion is present. Indeed, alternating distinct C–C bonds in the pyridyl groups and a short C–C distance (g) connecting the two pyridine rings, together with the planarity of the whole molecule, are characteristic of a reduced bipy [18a,21d]. The C–C(bipy) distances are quite similar in 2 but alternately differ by ∼ 0.04 Å in 3(py)2, suggesting the presence of more localized double bonds, as represented in Fig. 3. At last, the g bond length of 1.482(3) Å in 2 is close to that in free bipy [1.490(3) Å] and in f-metal complexes of neutral bipyridine, such as [Ce(C5Me5)2I(bipy)] [1.490(3) Å] [18a] or [UI3(bipy)2(py)] [1.49(2) Å] [18d], for example. This distance decreases by 0.06 Å in 3(py)2, and the value of 1.424(4) Å is similar to the corresponding C–C distance in [K(bipy)(ethylenediamine)]∞ or in complexes of the bipy•− anion, such as [U(C5Me5)(Cot)(bipy•−)] [1.427(3) Å] [21d] or [Sm(C5Me5)2(bipy•−)] [1.429(4) Å] [35]. This trend is reflected in the Th–N(bipy) distances which are in the range 2.687(2)–2.732(2) Å for 2, with a mean value of 2.703(17) Å longer by 0.15 Å than the corresponding mean Th–N distance in 3(py)2. A shortening of the U–N bond length by ca 0.2 Å was observed in U(IV) compounds of neutral and reduced bipyridine, such as in [U(OSiMe3)2I2(bipy)2] [2.646(5) and 2.670(5) Å] [18k] and [U(C5Me5)(Cot)(bipy−)] [2.429(2) and 2.468(2) Å] [21d]. All these structural features clearly demonstrate that 3(py)2 is a Th(IV) complex of the bipyridine radical anion rather than a true Th(III) species. At last, it is interesting to note that in the structure of 3(py)2, the potassium cation is not interacting with the bipy− ligand but with the Cot dianions. Because the bonding in f-element complexes is essentially ionic, the K+ ion is likely more prone to interact with the more accessible and charged Cot ligand than with the monoanionic bipy− in which the charge is delocalized over two aromatic cycles. Moreover, that the Cot dianion can bridge two different metals is now well established [33].
4 Conclusion
The greater reactivity of [Th(Cot)2] versus [U(Cot)2] was evidenced in its reaction with 2,2′-bipyridine, which gives [Th(Cot)2(bipy)] and provides a novel example of an unusual bent thorocene fragment. This bipy complex easily trapped one electron to give the anionic derivative [Th(Cot)2(bipy)][K(py)2], which forms a 1D polymer in the solid state. The comparison of the crystal structures of these two geometrically similar compounds reveals that the metal ion in [Th(Cot)2(bipy)]− is not Th3+ but Th4+ coordinated to the bidentate radical anion (bipy)−. Compounds 2 and 3(py)2 are rare examples of [An(Cot)2(L)2] complexes, and they are among the first molecular thorium species involving neutral or mono-anionic bipyridine ligands. These results also clearly demonstrate that the An4+ ion in [An(Cot)2] complexes is normally accessible to ligand coordination and can support one or even two ligands in its coordination sphere as it is the case within the analogous bis(C5Me5)2 series. Further work is in progress to explore the unique reactivity of thorocene and to explain theoretically the differences between uranium and thorium, which are likely attributable to the distinct covalent character of the An−Cot bond [17].
Acknowledgements
This work was financially supported by CEA and CNRS.