1 Introduction
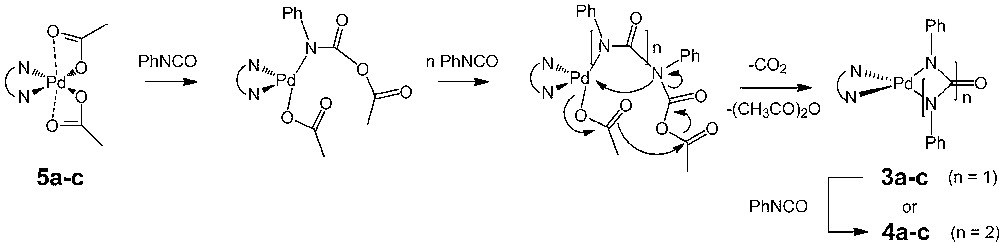
To date, the reactivity of isocyanates in the coordination sphere of various group-VIII metal centers is still a field only partially explored [1–3]. Such reactions can provide a very valuable access to various types of heterocycles and are thus especially attractive with respect to atom-economy considerations [4–7]. Moreover, they often bring some information about the possible deactivation pathways of catalytic processes involving isocyanate as a reactant or a product [8–10]. In this regard, we have previously shown that diimine-Pd(0) precursors catalytically trimerize arylisocyanates to form triarylisocyanurates (1, Scheme 1) [11], while some inactive palladacycles like 2, 3 or 4a [9,12], which possibly result from deactivation of the active species, are eventually also formed.

Cyclotrimer and palladacycles formed from diimine-Pd(0) complexes.
In the process of exploring the scope of this reaction, we have now investigated the reaction of PhNCO with various Pd(II)-phenanthroline precursors, like Pd(N–N)(OAc)2 complexes (5a-c), where N–N represents 1,10-phenanthroline (o-phen), 3,4,7,8-tetra-methylphenanthroline (tmphen), and 2,9-dimethylphenanthroline (dmphen), respectively. In a manner similar to the reaction with Pd(0) precursors, a reaction yielding the six-membered palladacycles 4a–c took place, providing thereby an easy access to these interesting compounds.
2 Results and discussion
When reacted with one equivalent of phenylisocyanate in dichloromethane at 20 °C, the bis-acetato diimine Pd(II) complex 5a [13,14] forms the known 6-membered palladacycle 4a [9,12] in ca. 30% yield after two days. In presence of an excess of PhNCO (20 equiv), this reaction becomes nearly quantitative, allowing isolation of ca. 95% of 4a after the same time. This reaction can also be performed at higher temperature in an aromatic solvent like toluene, 4a being then quantitatively isolated after 16 h (Scheme 2).
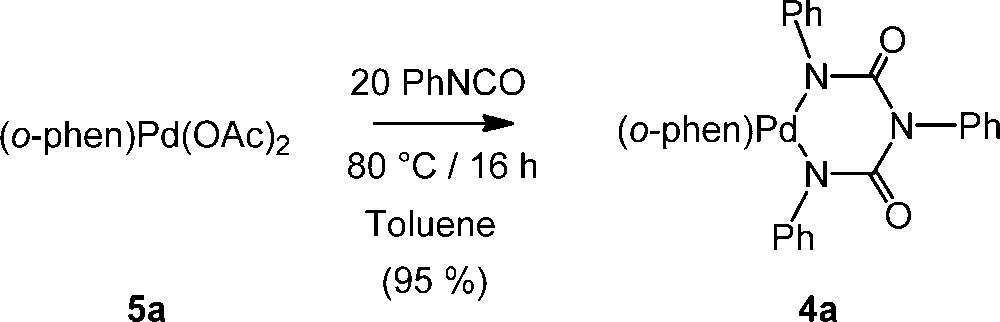
Reaction between 5a and PhNCO.
This reaction can also be performed with the analogue of 5a possessing a tmphen (5b) ligand instead of phen to give the corresponding palladacycle 4b in good yields. This new palladacycle was characterized by high-resolution mass spectrometry (HRMS), infrared and 1H NMR spectroscopies. In contrast, this reaction is not observed with other Pd(II) precursors such as Pd(o-phen)Cl2, Pd(OAc)2 or from an equimolar Pd(acac)2/o-phen mixture, pointing at the need to have a carboxylate anion and a diimine ligand simultaneously present in the medium for this reaction to proceed. Notably, we have checked that this reaction takes place with similar yield from a carefully dried complex 5a in anhydrous toluene, suggesting that traces of water are not involved in its mechanism.1
We then turned our attention toward the Pd(II) precursor 5c, presenting the more bulky dmphen ligand in place of o-phen [14], and with this sterically crowded Pd(II) complex, the reaction takes also place and a nearly complete conversion of 5c into 4c is observed in dichloromethane at ambient temperature after two days (Scheme 3). The new palladacycle 4c is the 2,9-dimethylated analogue of 4a. This complex was fully characterized, allowing notably the observation of the two strong carbonyl νCO stretching modes near 1645 and 1625 cm–1, diagnostic of the three carbonyl groups of this metallacycle [9]. Attempts to perform this reaction at 80 °C in toluene or other aromatic solvents were, however, not successful with partial decomposition of 5c taking place.
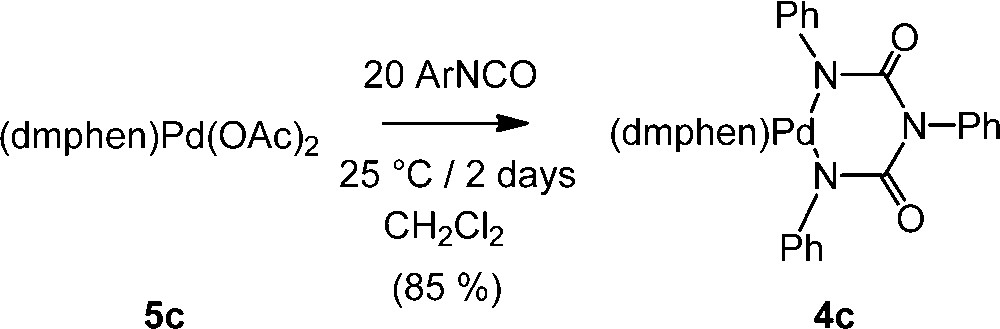
Reaction between 5c and PhNCO.
The steric pressure exerted by the two methyl groups is revealed by the X-ray structure presently obtained for 5c·OEt2·OH2 (Fig. 1).2 It results in a bending of ca. 30̊ of the phenanthroline core with the mean plane defined by the square planar coordination sphere around the Pd(II) atom. These crystallographic data are in line with those previously obtained for this and the related complex 5a, and clearly indicate that 5a–c are mononuclear species [14,15].
This reaction presents some analogy with the trimerization reaction of PhNCO catalyzed by “(N–N)Pd0” species previously studied by some of us [11]. However, the isocyanurate cyclotrimer 1 was never isolated as a side-product during any reaction of 5a with PhNCO, suggesting that “(o-phen)Pd0” is presently not transiently formed in the medium. Furthermore, it had been previously shown that the catalytic trimerization of PhNCO was not taking place with the sterically crowded 2,9-dimethylphenanthroline ligand. When this ligand was used, decomposition of the catalyst into metallic palladium was observed [11]. In contrast, the formation of palladacycle 4c takes readily place at ambient temperatures with the complex 5c. Thus, it is quite likely that the mechanistic pathway for the formation of 4a–c does not involve an elusive “(N–N)Pd0” intermediate. Actually, the reaction is observed only from dimine Pd(II) precursors having acetate ligands. A possible reaction mechanism should therefore involve the Pd(II) diimine center and the acetate ligand(s).
A tentative mechanistic proposal for this transformation is given in Scheme 4. The first step would involve the formal insertion of PhNCO into the Pd–O bond of the carboxylate ligand, possibly on a [Pd(N–N)(OAc)]+ intermediate, followed by further insertion of PhNCO into the resulting Pd–N bond (n = 2 or 3). These steps would be in line with the well-established insertion chemistry for this kind of cationic species [16]. It is indeed well known that on such complexes, the carboxylate ligands are labile and can be exchanged for other ligands [9,14]. Alternatively, this reaction sequence might be seen as a [Pd(N–N)(OAc)]+-assisted anionic oligomerization of PhNCO by the acetate ion, the latter acting as the nucleophile. This makes sense given the fact that simple acetate salts such as KOAc, in a manner similar to many other inorganic salts [17], are known to catalyze the cyclotrimerization of arylisocyanates [18]. Moreover, their activity as catalysts in this reaction has been shown to be strongly influenced by the nature of the associated cation [19]. However, this anionic oligomerization process would not ultimately lead to the cyclotrimer 1, but to the palladacycle 4a, due to the presence of the second acetate ligand on the nearby Pd(II) center. Indeed, the final steps would involve an intramolecular rearrangement taking place by elimination of acetic anhydride and carbon dioxide to form the metallacyclic core by ring closure. Such an acetate-based intramolecular rearrangement resemble the one proposed by Moiseev to rationalize the reduction of Pd(II) acetate clusters in presence of CO [20], except that the “CO” molecule is presently replaced by PhNCO. Then, depending on the number of inserted isocyanate molecules after ring closure, either 3a–c (n = 1) or 4a–c (n = 2) would be generated in the medium. Since we have previously clearly established that strained palladacycles such as 3a react readily with arylisocyanate to form 4a [9], this mechanism rationalizes the selective formation of the 6-membered palladacycles 4a–c in the presence of an excess of PhNCO.3 Also, based on literature reports [14,22], the acetate ligand should be more labile in 5c than in 5a. If the first insertion of isocyanate in the Pd–OAc bond in the former complex is rate determining, the reaction might take place as fast with 5c than with 5a, in line with our preliminary observations.
3 Conclusions
In conclusion, we report here a novel and efficient reaction toward the six-membered palladacycles 4a–c from air-stable phenanthroline Pd(II) acetate complexes and phenylisocyanate. While revealing that the particular complexes 5a–c are reactive toward arylisocyanates, this contribution clearly shows that other synthetic routes than these previously evidenced from [(N–N)Pd0] intermediates are possible to obtain palladacycles such as 2–4. Thus, such species might also be directly formed from Pd(II) catalyst precursors when carboxylate ligands are present in the medium. This observation is particularly important in the context of Pd-catalyzed reductive carbonylations of nitroaromatics, because (Pd(II)/diimine/carboxylic acid) catalytic systems are among the most active ones [10] and because the formation of 2–4 and related palladacycles [23] has been proposed to be at the origin of the rapid deactivation of these catalytic systems [8,9], especially when these reactions are performed in the absence of alcohol or amine able to trap the isocyanate formed [24]. The scope and reaction mechanism of this new reaction is currently under investigation.
4 Experimental
4.1 General
The solvents were dried and distilled prior to use and the reaction was performed under inert (Ar) atmospheres in deaerated solvents. Unless otherwise specified, all reagents were purchased from commercial suppliers and used without further purification. Infrared spectra were recorded on a Bruker IFS28 spectrometer (400–4000 cm–1). 1H NMR spectra were obtained using Bruker SY 200 (200 MHz) or SY 400 (400 MHz) Fourier Transform spectrometers. Chemical shifts are given in parts per million relative to tetramethylsilane (TMS) for 1H spectra. Mass spectral studies and elemental analyses were entrusted to the corresponding services of the Centre régional de mesures physiques de l’Ouest (CRMPO). The complexes 5a–c were synthesized according to previously reported procedures [13,14] (see also supporting information).
4.2 Reactions of Pd(N–N)(OAc)2 and PhNCO
To 100 mg of Pd(phen)(OAc)2 (0.24 mmol) were added 600 mg of phenyl isocyanate (20 equiv) dissolved in 15 mL of toluene. The pale-yellow suspension was subsequently heated at 80 °C under stirring. After 16 h, an abundant orange-yellow suspension had formed in the reaction medium. The medium was cooled and 10 mL of ethanol were added to neutralize the excess of isocyanate. The suspension was then decanted and washed with several fractions of diethylether, yielding 140 mg of a yellow-orange solid after subsequent vacuum-drying. This solid is identified as palladacycle 4a by comparison with authentic samples (95%).
To 50 mg of Pd(tmphen)(OAc)2 (0.11 mmol) were added 630 mg of phenyl isocyanate (50 equiv) dissolved in 15 mL of toluene. The pale-yellow suspension was subsequently heated at 80 °C under stirring. After 16 h, the dark-red reaction medium was cooled and 1 mL of EtOH was added, followed by addition of an excess of diethylether. The suspension was then decanted and washed with several fractions of diethylether, yielding 39 mg of a brown–orange solid after vacuum-drying. This solid is identified as palladacycle 4b (54%). Color: brown–orange. HRMS (positive ESI, CH2Cl2/MeOH, m/Z) z 672.1578 [M+H]+, m/z calculated for [C36H32N5O2Pd] 672.1591. FT-IR (KBr, ν, cm–1) 1642 (s, C=O), 1623 (s, C=O). 1H NMR (200 MHz, CDCl3, δ, ppm) 8.53 (d, 3JHH = 8 Hz, 4H, HPh), 8.05 (s, 24H, Hphen), 7.88 (s, 2H, Hphen), 7.53 (t, 3JHH = 8 Hz, 4H, HPh), 7.36–6.98 (m, 7H, HPh), 2.68 (s, 6H, CH3/phen), 2.20 (s, 6H, CH3/phen).
To 100 mg of Pd(dmphen)(OAc)2 (0.23 mmol) dissolved in 15 mL of CH2Cl2 were added 550 mg of phenylisocyanate (ca. 20 equiv). The orange solution was stirred during two days at ambient temperature. The solution was filtrated and diethylether was added to precipitate the title complex as an orange solid. It was collected, washed with several fractions of diethylether and dried in vacuo, yielding 130 mg of the title palladacycle (88%). Color: orange. Dec. Pt: 180 ± 10 °C. HRMS (positive ESI, CH2Cl2, m/Z) 644.1291 [M+H]+, m/z calculated for [C34H28N5O2Pd] 644.1278. Anal. calculated for C34H27N5O2Pd·H2O: C, 61.68; H, 4.42; N, 10.58; found: C, 61.67; H, 4.48; N, 10.52. FT–IR (KBr/Nujol, ν, cm–1) 1645 (s, C=O), 1625 (s, C=O). 1H NMR (200 MHz, CD2Cl2, δ, ppm) 8.26 (d, 3JHH = 8 Hz, 2H, Hphen), 8.01 (d, 3JHH = 7 Hz, 4H, HPh), 7.90 (s, 2H, Hphen), 7.69 (d, 3JHH = 8 Hz, 2H, Hphen), 7.49 (t, 3JHH = 8 Hz, 2H, HPh), 7.37 (t, 3JHH = 8 Hz, 4H, HPh), 7.26 (d, 3JHH = 8 Hz, 2H, HPh), 6.89 (t, 3JHH = 7 Hz, 4H, HPh), 6.69 (t, 3JHH = 7 Hz, 1 H, HPh), 2.72 (s, 6H, CH3/phen).
4.3 X-ray crystallography
Diffraction data frames for 5c were collected using a CCD Saphire 3 Xcalibur apparatus using the graphite monochromatized Mo Kα radiation (λ = 0.71073 Å). The cell parameters were obtained with Denzo and Scalepack [25], with 10 frames (psi rotation: 1̊ per frame). The data collection leads to 4235 independent reflections, from which 3338 with I > 2.0 σ(I) [26]. The structures were solved by direct methods using SIR97 program [27], and then refined with full-matrix least-square methods based on F2 (SHELX-97) [28]. The contribution of the disordered solvents (i.e. one molecule of diethylether and one molecule of water) to the calculated structure factors was removed using the SQUEEZE option in PLATON. Atomic scattering factors were taken from the International tables for X-ray crystallography (1992) [29]. Ortep views were realized with PLATON98 [30].
Acknowledgements
We thank the CNRS for financial support and “Rhône-Poulenc Recherches” for a PhD grant (F.P.) when this work was initiated under the supervision of J.A. Osborn.
Appendix A Supplementary data
Synthesis and characterization of 5a-d. CCDC-221834 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via: www.ccdc.cam.ac.uk/data_request.cif.
1 We have stated that Pd(II) complexes 5a–c often give rise to quite stable solvates with water molecules. A water-free sample of 5a could however be obtained after reacting this complex with acetic anhydride in nitrobenzene (12 h, 80 °C) followed by recrystallization with Et2O from nitrobenzene.
2 This solvated complex presents nearly identical crystallographic parameters than two different solvates of 5c previously reported.[14] Crystallographic data after refinement without solvate(s): PdC18H18N2O4, M = 524.88, monoclinic, space group C 2/c, a = 18.3528(5) Å, b = 18.1223(6) Å, c = 13.4071(4) Å, β = 119.459(1) ̊, U = 3882.6(2) Å3, Z = 4, T = 120(2) K, Dc = 1.481 g cm–3. The final wR(F2) was 0.073 (all data). CCDC deposition No.: CCDC 221834.

3 With n = 0, a Pd-imido diimine intermediate would be formed rather than a metallacycle. While such kind of complex has recently been claimed to play a determining role in the catalytic carbonylation of nitroaromatics [21], we did not consider the possibility of its intermediacy here. However, based on the available data [1,9], such a complex should also transform quantitatively into 4a–c in presence of excess PhNCO.