

1 Introduction
Prussian blue analogues (PBAs) form a wide class of inorganic polymers exhibiting numerous appealing electronic and chemical properties, including room-temperature magnetic properties [1,2], large porous volumes (suitable for gas storage applications) [3–5], electrochemical properties for alkali cation-based batteries [6,7], and electronically switchable properties [8–10]. The understanding of the physical and chemical phenomena occurring at the atomic scale in PBAs is needed to finely tune up these promising properties. Infrared (IR) spectroscopy has recently been evidenced as a well-adapted probe of the electronic and structural properties of PBAs at the molecular level [11]. However, no comprehensive assignment of the IR spectrum of PBAs was available—to date—in the literature.
We demonstrate herein the relevance of the IR approach to the probing of the electronic and structural properties of PBAs. A comprehensive assignment of the IR spectrum of PBAs is performed based on the electronically switchable series of cobalt–iron PBAs [12,13]. Both the far-IR region and the spectral range associated with the ν{OH} of the water molecules of PBAs are discussed. Using these assignments, examples of the use of IR spectroscopy as an accurate tool to investigate PBAs are proposed, including monitoring of the electronic transition, discussion of the chemical disorder, or probing of the interaction between the alkali cations and the bimetallic network.
2 Experimental
The syntheses of K0CoFe, Cs0.7CoFe, Na2CoFe, Rb2CoFe and Cs2CoFe have been already described elsewhere [12,14]. IR/THz measurements were carried out at the AILES beamline at synchrotron SOLEIL (France). The synchrotron was operating in the equal filling mode with 416 approximately equally filled and spaced electron bunches. The average storage beam electron current for the present result was 400 mA. The IR/THz experimental set-up has already been described elsewhere [15,16]. All THz measurements were performed in the transmission mode using a Bruker IFS 125 Fourier transform interferometer fitted with 6 μm Mylar/Si multilayer beam splitter and a liquid-helium-cooled Si bolometer detector, and operated at 1-cm−1 resolution. The MIR measurements exploited the interferometer fitted with a KBr beamsplitter combined with a MCT detector. The Fourier transform spectrometer was evacuated down to a 2 × 10−5 mbar pressure (to minimize residual H2O and CO2 absorption in the spectrometer). The liquid absorption cell used for these experiments was filled with sample powder dispersed in Nujol and held between two diamond windows. For the FIR spectra, the detector was fitted with a 200–600-cm−1 cold optical filter and the spectrometer with a 12.5-mm entrance aperture, but the SR effective source diameter results in beam diameter that fulfills the resolution criterion. All spectra result from the averaging of 400 scans measured with a mobile mirror speed of 2.5 cm·s−1. In order to measure the absorption spectra at well-controlled temperatures, the liquid cell was mounted on a cold head controller by a close cycle cryostat (pulse tube from CryoMech). This set-up allowed the sample temperature to be controlled within 1 K. The transmission spectra were obtained by dividing the signal (I) by the signal transmitted through pure Nujol (I0). All IR spectra are presented in absorbance (A = ln(I/I0)) as a function of the incident wavenumbers ω.
3 Results and discussion
3.1 Description of PBAs
PBAs are coordination polymers obtained in aqueous solution from the substitution of the water molecules in [M(OH2)6]k+ complexes by the isocyanide ligands from [M′(CN)6]j− complexes (where M and M′ are transition metal ions). The resulting MN≡CM′ linkages form a face-centered cubic lattice [17] that may exhibit vacancies in M′(CN)6 units (Fig. 1). In the vicinity of these vacancies, water molecules are coordinated to the M cations. Additional alkali cations and zeolite water molecules can be inserted into the structure. The general formula of PBAs is AxM4[M′(CN)6]α□4−α·nH2O (where A+ is an alkali cation and □ is a M′(CN)6 vacancy; α = 4(k + (x/4))/j), called AxMM′ in the following. Depending on the nature of the A, M and M′ cations, the formers are known to interact with the bimetallic network in some PBAs [13].
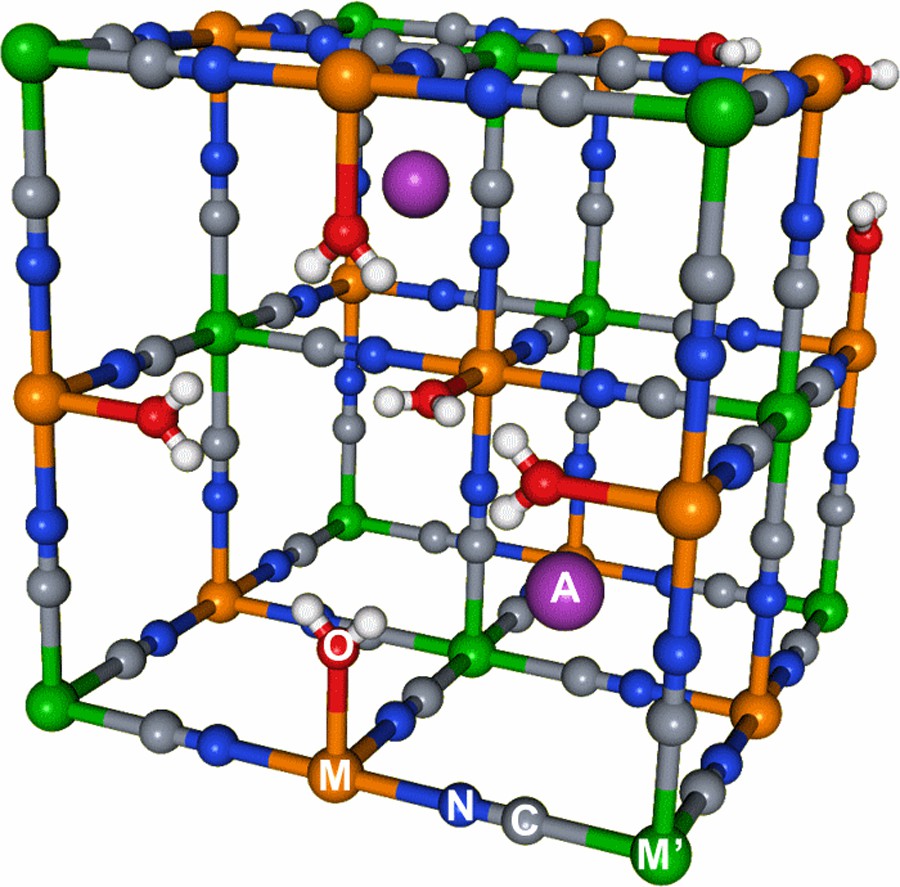
Scheme of the unit cell of a Prussian blue analogue. Colors: M (orange), M′ (green), C (grey), N (blue), O (red), H (white), A (purple). For interpretation of references to color, see the online version of this article.
The assignment of the IR vibration bands of PBAs has been performed thanks to a series of cobalt–iron PBAs (AxCoFe) where k = 2 and j = 3. The general formula of AxCoFe is therefore given by AxCo4[Fe(CN)6](8+x)/4□(4−x)/3·nH2O. Thus, the stoichiometry of AxCoFe is completely defined by the amount x of alkali cations inserted in the structure. The AxCoFe PBAs can exhibit two different CoII(HS)FeIII(LS) and CoIII(LS)FeII(LS) (HS: high spin; LS: low spin) electronic states [18] (called CoIIFeIII and CoIIIFeII in the following). However, as the amounts of cobalt and iron cations per unit cell are not the same, part of cobalt cations cannot undergo the CoIIFeIII ↔ CoIIIFeII electronic transition. Consequently, the chemical formula of AxCoFe PBAs can be rephrased as AxCoII4[FeIII(CN)6](8+x)/3□(4−x)/3·nH2O (in the CoIIFeIII state) and AxCoIII(8+x)/3CoII(4−x)/3[FeII(CN)6](8+x)/3□(4−x)/3·nH2O (in the CoIIIFeII state), taking into account the small amount of residual CoII cations in the CoIIIFeII state.
According to the chemical composition and the structure of PBAs, their IR spectrum is expected to exhibit the following vibration bands:
- • the ν{C≡N} vibration band, related to the cyanide bridges;
- • the ν{MN}, ν{MO} and ν{M′C} vibration bands, related to the metal-to-ligand bonds;
- • the ν{OH} vibration band, related to the water molecules of the system (both zeolite water molecules and water molecules bound to the M cations at the M′(CN)6 vacancies).
3.2 Assignment of the ν{C≡N} vibration band
The ν{C≡N} vibration band is located in the 2100–2200-cm−1 spectral range. As the cyanide bridge is extremely sensitive to its environment, including the oxidation state and the spin state of the M and M′ cations, the ν{C≡N} band has already been extensively used to characterize the electronic state of switchable PBAs [19]. In the case of the AxCoFe PBAs, the ν{C≡N} vibration band exhibits a maximum located at 2120 cm−1 in the CoIIIFeII state, characteristic of the cyanide bridge in the CoIIIN≡CFeII linkages [19,20]. In the CoIIFeIII state, the maximum, located at 2160 cm−1, is characteristic of the cyanide bridge in the CoIIN≡CFeIII linkages [19,20]. The several minor contributions located between 2100 and 2120 cm−1 have been assigned to residual CoIIN≡CFeII linkages, surface non-bridging cyanides or residual CoIIIN≡CFeII linkages [21]. The IR spectrum of the thermally switchable Na2CoFe PBA [19,22] in both the CoIIFeIII and CoIIIFeII states is shown in Fig. 2. While the ν{C≡N} band shall contain a tremendous amount of information concerning PBAs (presence of minority species, interaction between the alkali cations and the bimetallic network, etc.), hardly obtainable through other techniques, the superimposition of several contributions for this vibration band over a very small spectral range results in a mutual hindering of this information. Consequently, the metal-to-ligand vibrations bands were investigated in order to unravel these various contributions to the electronic properties of PBAs.
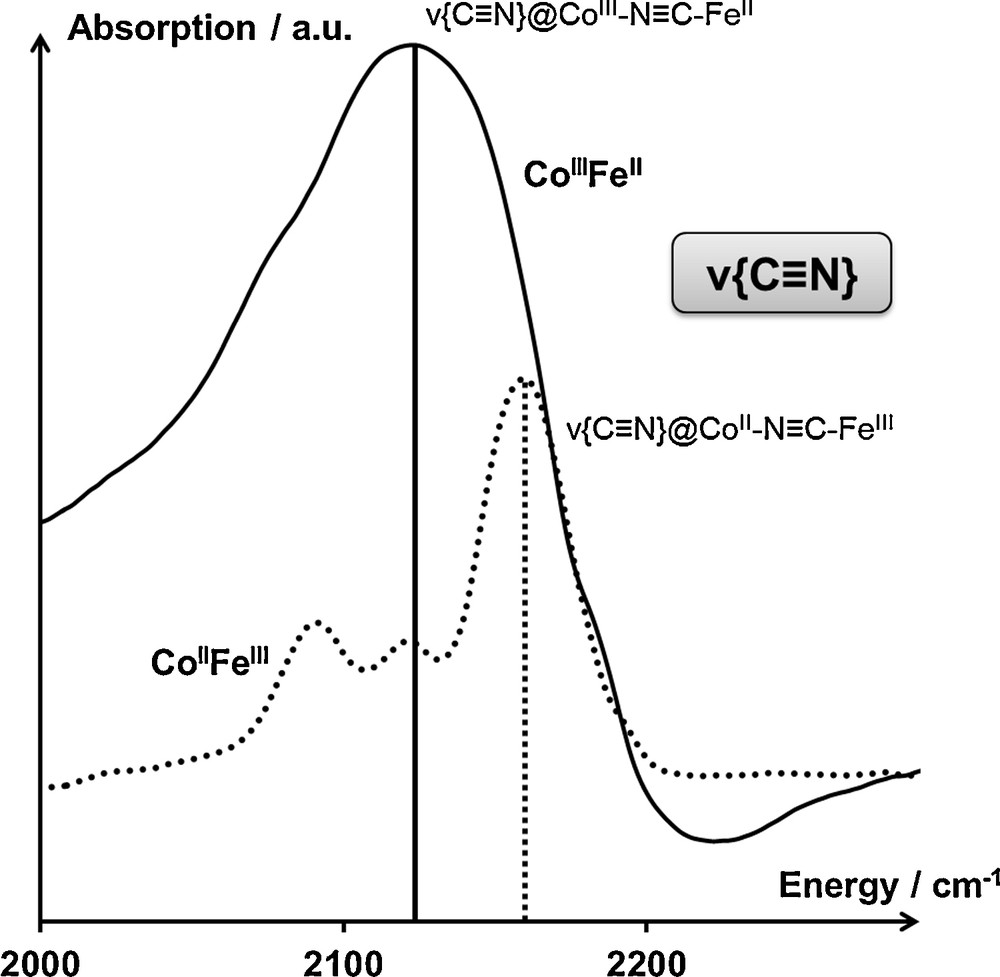
Infrared spectrum of Na2CoFe in the spectral range associated with the ν{C≡N} vibration band, both in the CoIIFeIII (T = 300 K) and CoIIIFeII (T = 200 K) electronic states. Assignments are detailed in the main text.
3.3 Assignment of the ν{ML} vibration bands
The assignment of the ν{ML} vibration bands has been performed based on the CoIIFeIII ↔ CoIIIFeII thermally activated transition of Na2CoFe [19,22] (Fig. 3). The IR spectrum of Na2CoFe measured at different temperatures (200 K and 300 K) exhibits two pairs of vibration bands, coupled during the electronic transition, located respectively in the 150–350-cm−1 and 400–600-cm−1 spectral ranges. According to the literature dealing with the [M(NH3)6]k+ (resp. [M′(CN)6]j−) complexes [23], the vibration bands in the 150–350-cm−1 (resp. 400–600 cm−1) spectral range have been assigned to the CoN (resp. FeC) bonds.
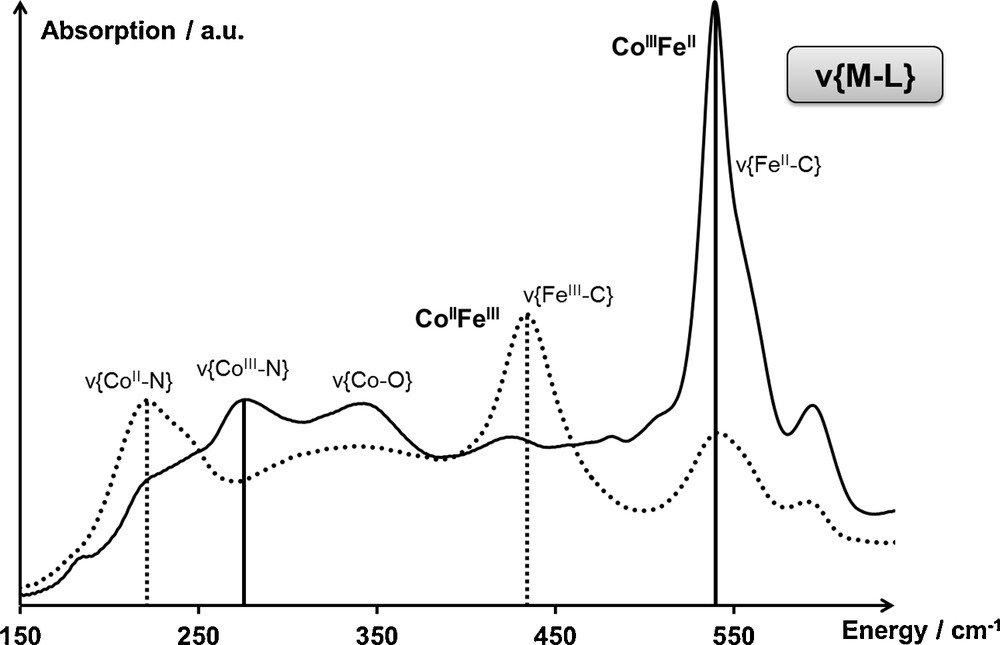
Infrared spectrum of Na2CoFe in the spectral range associated with the ν{ML} vibration band, both in the CoIIFeIII (T = 300 K) and CoIIIFeII (T = 200 K) electronic states. Assignments are detailed in the main text.
Based on the CoIIFeIII electronic state of Na2CoFe at T = 300 K, the band located at 220 cm−1 has been assigned to the CoIIN bonds and the one located at 430 cm−1 has been assigned to the FeIIIC bonds. Similarly, based on the CoIIIFeII electronic state of Na2CoFe at T = 200 K, the band located at 270 cm−1 has been assigned to the CoIIIN bond and the band located at 540 cm−1 has been assigned to the FeIIC bond. Finally, the broad bands located in the 250–400 cm−1 spectral range have been assigned to the CoII/IIIO bonds, according to the literature dealing with the [M(OH2)6]k+ complexes [23].
These assignments are in agreement with the vibration bands observed for the free [M′(CN)6]j− complex [23]; for instance, the displacement of the ν{FeIIC} vibration band from 585 cm−1 in [Fe(CN)6]4− to 540 cm−1 in the CoIIIFeII state of Na2CoFe is consistent with the depletion of the electronic density on the Fe(CN)6 entity due to the coordination of the isocyanide ligands to the cobalt cations [23].
3.4 Assignment of the ν{OH} vibration bands
The spectral range associated with the ν{OH} vibration band in PBAs comprises two distinct regions:
- • from 3000 to 3500 cm−1, the IR spectrum exhibits very broad bands associated with water molecules involved in a hydrogen-bonded network;
- • from 3550 to 3700 cm−1, the IR spectrum exhibits one or several very sharp bands associated with water molecules that are not involved in a hydrogen-bonded network.
The number of the sharp bands in the 3550–3700-cm−1 spectral range may vary depending on the nature of the PBA (supplementary data). According to their position in energy, comprised between 3590 cm−1 (water dimers [24]) and 3755 cm−1 (isolated water molecules [25]), the bands located in the 3600–3700 cm−1 range have been assigned to water molecules bound to metallic cations (either transition metal cations M or alkali cations A). This assignment can be further refined when considering the electronic transition of the Na2CoFe PBA (Fig. 4).
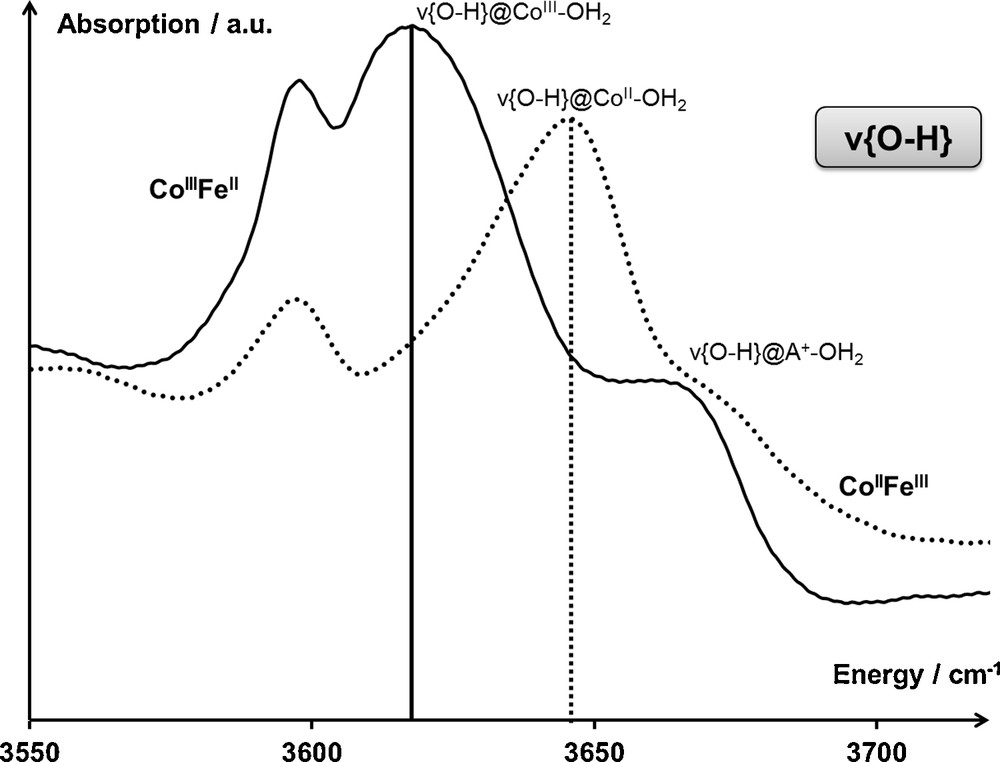
Infrared spectrum of Na2CoFe in the spectral range associated with the ν{OH} vibration band of water molecules that are not involved in a hydrogen-bonded network, both in the CoIIFeIII (T = 300 K) and CoIIIFeII (T = 200 K) electronic states. Assignments are detailed in the main text.
Amongst the three vibration bands located in the 3600–3700-cm−1 range in Na2CoFe, the two located at a lower energy are evolving through the CoIIFeIII ↔ CoIIIFeII electronic transition. Consequently, these two bands have been assigned to water molecules bound to the cobalt cations at the Fe(CN)6 vacancies. Based on the CoIIFeIII electronic state of Na2CoFe at T = 300 K, the band located at 3645 cm−1 has been assigned to the ν{OH} vibration of water molecules bound to CoII cations; similarly, based on the CoIIIFeII electronic state of Na2CoFe at T = 200 K, the band located at 3620 cm−1 has been assigned to the ν{OH} vibration of water molecules bound to CoIII cations. This assignment is consistent with the water-to-bimetallic network partial charge transfer relying on the mostly σ character of the water-to-metal bond: in the CoIIFeIII electronic state, the depletion of the electronic density on a water molecule bound to the CoII cation is rather small, which results in a rather strong OH bond (ν{OH} = 3645 cm−1); in the CoIIIFeII electronic state, the depletion of the electronic density on a water molecule bound to the CoIII cation is much more important, which results in a weaker OH bond (ν{OH} = 3620 cm−1).
The higher energy band has been assigned to water molecules bound to alkali cations. The appearance and the intensity of this latter vibration band are highly sensitive to the nature and the amount of alkali cations per unit cell (supplementary data).
This assignment of the IR spectrum of PBAs over the whole 100–4000-cm−1 spectral range allows the fine investigation of the structure of PBAs (presence of vacancies, position of the alkali cation in the lattice, etc.) and the alkali-cyanide interaction [13]. The following sections aim at demonstrating the relevance of the IR spectroscopy in the study of such properties of PBAs, which cannot be probed by other techniques.
3.5 Signatures of the M′(CN)6 vacancies
In the CoIIFeIII electronic state, the chemical formula of AxCoFe PBAs can be rephrased as AxCoII4[FeIII(CN)6](8+x)/3□(4−x)/3·nH2O. Consequently, the mean composition of the coordination sphere of the FeIII cations is always given by FeIII(C≡NCoII)6, whatever the amount x of alkali cations inserted in the structure. A single type of ν{FeIIIC} vibration band, associated with the CoIIN≡CFeIII linkages, is therefore expected for the AxCoFe PBAs in the CoIIFeIII electronic state.
The IR spectrum of the AxCoFe PBAs in the CoIIFeIII electronic state in the spectral range associated with the ν{FeIIIC} vibration band shows a single contribution, whichever the nature and the amount x of inserted alkali cations (Fig. 5). This observation is consistent with the expected single contribution to this band for the AxCoFe PBAs in the CoIIFeIII electronic state.
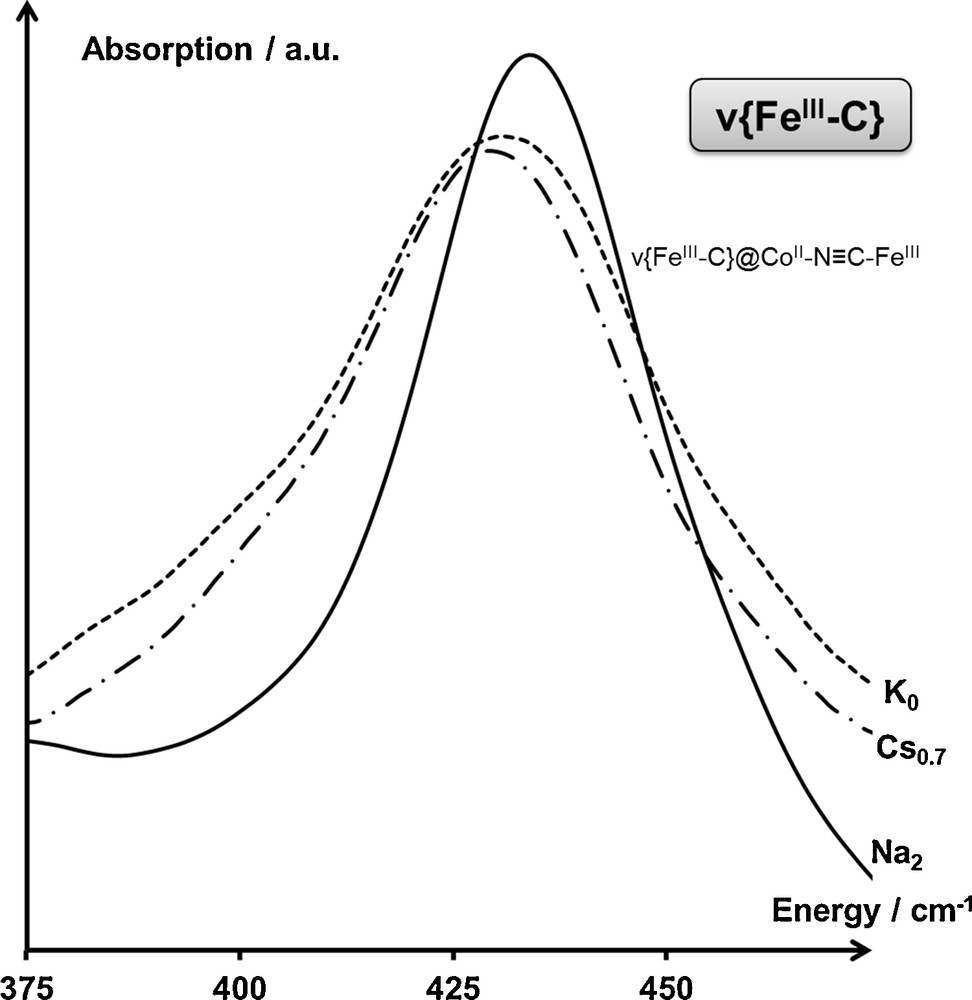
Infrared spectrum of the K0CoFe (---), Cs0.7CoFe (—·—), Na2CoFe (—) PBAs in the CoIIFeIII electronic state, in the spectral range associated with the ν{FeIIIC} vibration band, at T = 300 K. Assignments are detailed in the main text.
The situation is however very different for the CoIIIFeII electronic state: according to the AxCoIII(8+x)/3CoII(4−x)/3[FeII (CN)6](8+x)/3□(4−x)/3·nH2O chemical formula of the AxCoFe PBAs in the CoIIIFeII state, each octahedral FeII cation exhibits a mean FeII(C≡NCoIII)(8+x)/2(C≡NCoII)(4−x)/2 coordination sphere. While the presence of residual CoII cations can be monitored by magnetic measurements, the subsequent CoIIN≡CFeII linkages are hardly measurable by IR spectroscopy in the ν{C≡N} spectral range due to:
- • the small amount of these CoIIN≡CFeII linkages in CoIIIFeII PBAs;
- • the superimposition of numerous contributions to the IR spectrum in the ν{C≡N} region.
In Na2CoFe (x = 2), the mean coordination sphere of the iron cations depends on the electronic state of the system: in the CoIIFeIII state, the mean coordination sphere of FeIII is FeIII(C≡NCoII)6, while in the CoIIIFeII state, the mean coordination sphere of FeII is FeII(C≡NCoIII)5(C≡NCoII)1. In the CoIIFeIII state, the IR spectrum of Na2CoFe (Fig. 6a) exhibits a single symmetrical band (ν{FeIIIC} = 433 cm−1); in the CoIIIFeII state, the IR spectrum of Na2CoFe (Fig. 6b) exhibits a well-defined main contribution (ν{FeIIC} = 539 cm−1) and a broad shoulder at higher energy. These observations are consistent with the anticipated coordination spheres for the iron cations: the single contribution to the ν{FeIIIC} vibration band can be assigned to the FeIIIC bond in CoIIN≡CFeIII linkages; the two contributions to the ν{FeIIC} vibration band can be assigned to the FeIIC bonds in CoIIIN≡CFeII and CoIIN≡CFeII linkages. According to:
- • the relative intensity of the two contributions to the ν{FeIIC} vibration band;
- • the broadness of the contribution at higher energy, the main contribution has been assigned to the FeIIC in the majority CoIIIN≡CFeII linkages, while the higher energy contribution has been assigned to the FeIIC in the minority CoIIN≡CFeII linkages.
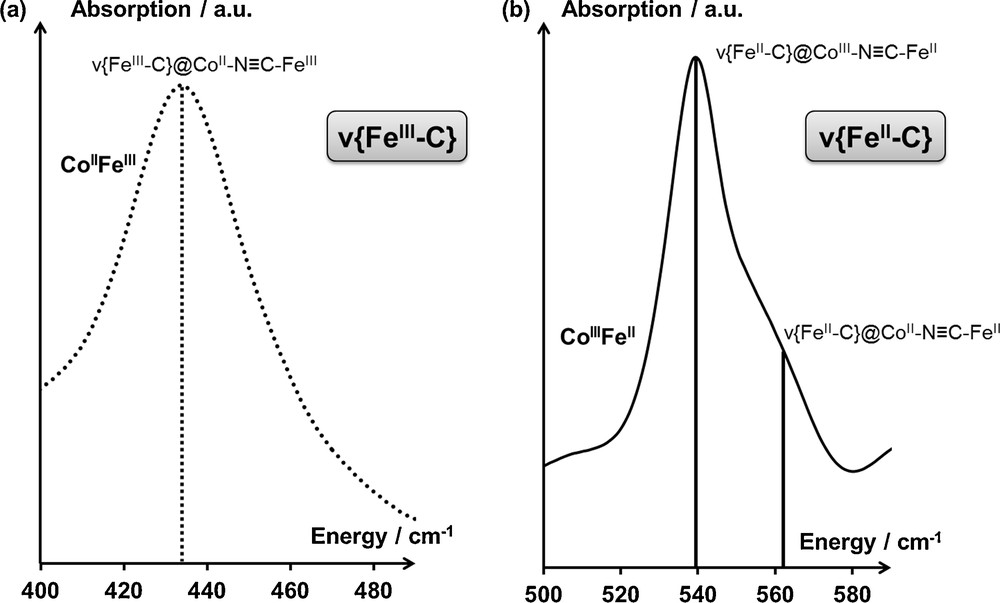
Infrared spectrum of Na2CoFe in the spectral range associated with the ν{FeC} vibration band, (a) in the CoIIFeIII (T = 300 K) electronic state and, (b) in the CoIIIFeII (T = 200 K) electronic state. Assignments are detailed in the main text.
The broad width of the higher energy shoulder is consistent with the mismatch between the CoIIN≡CFeII linkage and the surrounding mostly CoIIIFeII environment, resulting in some structural disorder.
This study highlights the relevance of the IR spectroscopy in the investigation of the different CoN≡CFe linkages in AxCoFe PBAs.
3.6 Study of the alkali–cyanide interaction
The existence of an interaction between the alkali cations and the bimetallic network has already been evidenced in some AxCoFe PBAs [13]. This interaction, whose nature is still debated, may result in the modulation of the relative stability of the CoIIFeIII and CoIIIFeII electronic states, as evidenced, for instance, in the A2CoFe series: while the Rb2CoFe and Cs2CoFe PBAs always exhibit a CoIIIFeII ground state [26], Na2CoFe shows a thermal transition between the CoIIFeIII and CoIIIFeII states around T = 250 K [19,22]. According to the nucleophilic nature of the cyanide bridge, the interaction between the alkali cations and the bimetallic network is expected to involve the electron-rich bridging cyanide ligands. However, no direct signature of this interaction has been, to date, proposed in the literature.
In the A2CoFe series of PBAs, all the compounds exhibit the same {Co4[Fe(CN)6]3.3□0.7(OH2)4} bimetallic network. In the CoIIIFeII state, the cobalt cations of these PBAs share a common CoIII(N≡CFeII)5(OH2)1 mean coordination sphere. The IR spectra of Rb2CoFe and Cs2CoFe in the CoIIIFeII state (Fig. 7) exhibit a single contribution to the ν{CoIIIN} vibration band, which has therefore been assigned to the CoIIIN in the CoIIIN≡CFeII linkages. However, the IR spectrum of Na2CoFe in the CoIIIFeII state (Fig. 7) shows two distinct contributions for the ν{CoIIIN} vibration band. The nature of the alkali cation being the sole difference between these A2CoFe PBAs, this splitting of the ν{CoIIIN} vibration band is therefore due to the different interactions between the alkali cations and the bimetallic network.
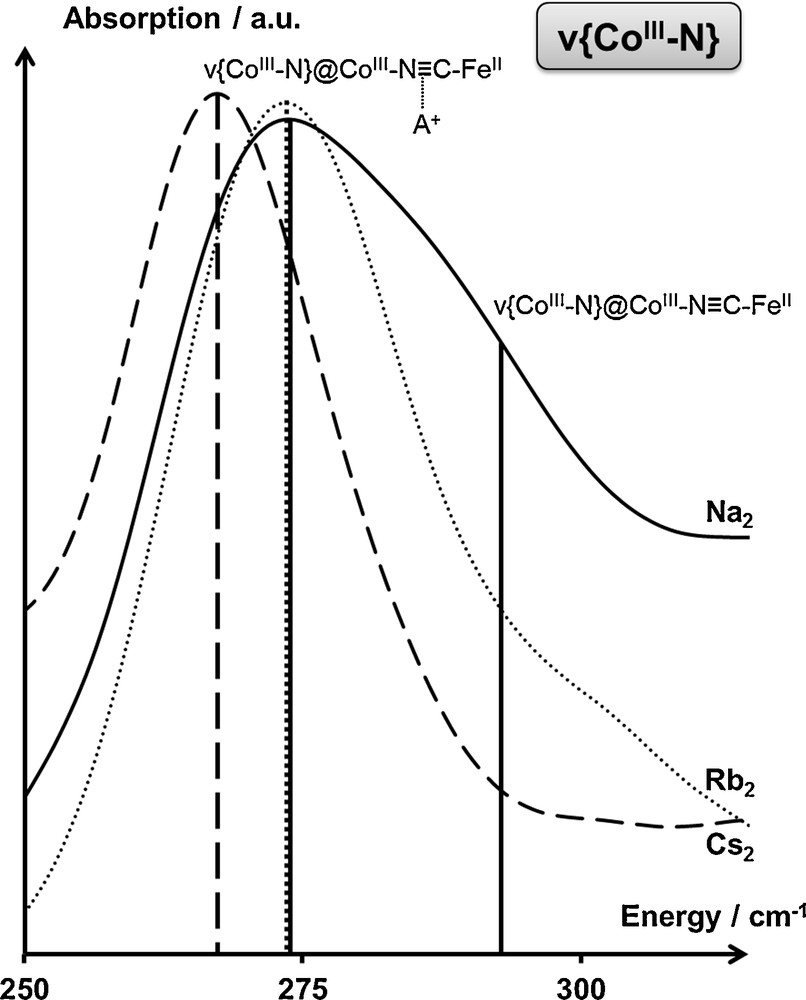
Infrared spectrum of the Na2CoFe (—), Rb2CoFe () and Cs2CoFe (— — —) PBAs in the CoIIIFeII state (T = 200 K) in the ν{CoIIIN} spectral range. Assignments are detailed in the main text.
The presence of two contributions for the ν{CoIIIN} vibration band in Na2CoFe reveals two chemically non-equivalent CoIIIN bonds, which have been assigned to CoIIIN≡CFeII linkages interacting and non-interacting with an alkali cation [11]. Thus, the difference in energy between these two contributions is a probe of the strength of the alkali–cyanide interaction.
This assignment can be further confirmed by considering the CsxCoFe series of PBAs. In the case of large alkali cations standing at the center of the octants of the lattice (Fig. 1), each alkali cation can interact with a maximum of 12 cyanide bridges. In a given AxCoFe series, each unit cell comprises 2 × (8 + x) cyanide bridges; therefore a maximum of 2x × (8 + x) cyanide bridges per unit cell are interacting with an alkali cation. Consequently, if x ≥ 1, each cyanide bridge is interacting with an alkali cation, and the resulting ν{CoN} vibration band should exhibit a single contribution. On the contrary, in the case of AxCoFe PBAs with low amounts of alkali cations per unit cell (x < 1), two contributions are expected for the ν{CoN} vibration band, one corresponding to the CoN in CoN≡CFe linkages that are not interacting with an alkali cation, and the other corresponding to CoN≡CFe linkages interacting with an alkali cation. Such assumption can be confirmed in the CsxCoFe series: while the Cs0.7CoFe PBA (x < 1) shows a splitting of the ν{CoIIN} vibration band in the CoIIFeIII electronic state (Fig. 8), no splitting of the ν{CoIIIN} vibration band is observed in the CoIIIFeII electronic state of Cs2CoFe (x ≥ 1) (Fig. 7). Furthermore, no splitting of the ν{CoIIN} vibration band is observed in the case of the alkali-free K0CoFe PBA (Fig. 8).
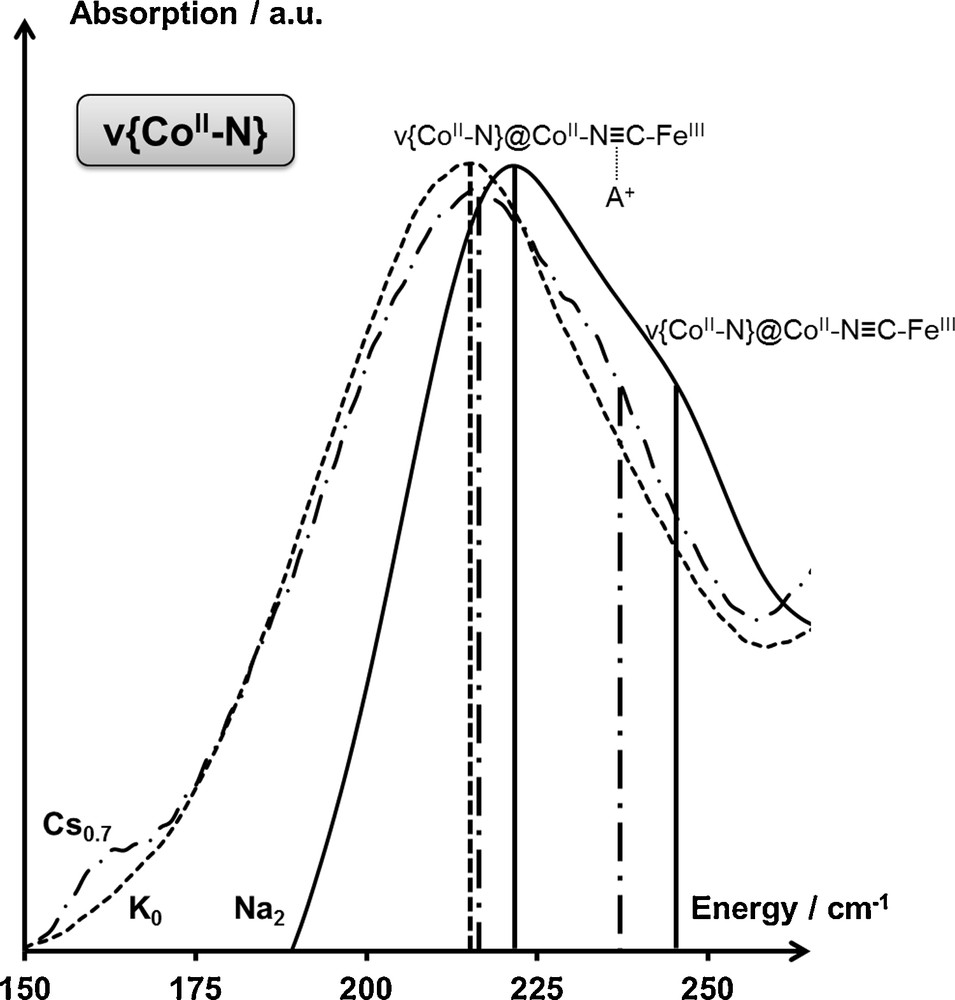
Infrared spectrum of the K0CoFe (---), Cs0.7CoFe (—·—) and Na2CoFe (—) Prussian blue analogues in the CoIIFeIII state (T = 300 K) in the spectral range associated with the ν{CoIIN} vibration band. Assignments are detailed in the main text.
3.7 Positioning of the alkali cation
IR study of the alkali-cyanide interaction taking place in AxCoFe PBAs can also bring some insight into the much debated question of the position of the alkali cation in the lattice. While the initial description of PBAs locates the alkali cations at the center of the octants of the lattice [17], recent studies tend to question this positioning [11,27]. Indeed, according to the distance between the center of the octants and the cyanide bridges, this positioning seems unsuitable for smaller alkali cations. Consequently, in the case of small alkali cations, a splitting of the ν{CoN} vibration band is anticipated even for AxCoFe PBAs with an important amount of alkali cations per unit cell. Experimentally, two distinct contributions to the ν{CoN} vibration band are observed for the Na2CoFe PBA, for which both the ν{CoIIN} vibration band (in the CoIIFeIII electronic state; Fig. 8) and the ν{CoIIIN} vibration band (in the CoIIIFeII electronic state; Fig. 7), corresponding to the CoN bonds implied either in the CoN≡CFe linkages interacting with an alkali cation, or in the CoN≡CFe linkages that are not interacting with an alkali cation. This splitting of the ν{CoN} vibration band in Na2CoFe indicates that the sodium cation does not stand at the center of the octants of the lattice. Such results demonstrate that IR spectroscopy can bring new insights into the intimate structure of PBAs, as such information cannot be inferred from standard X-ray diffraction studies of PBAs containing small, disordered alkali cations. Further refinement of the position(s) of the alkali cations in the unit cell of PBAs and highlighting of possible displacements of the alkali cations in the unit cell are in progress.
Finally, the impact of the interaction between the alkali cation and the bimetallic network on the metal–ligand bonds can also be pointed out in the Cs2CoFe PBA: while no splitting of the ν{CoIIIN} vibration band is observed for this system in the CoIIIFeII state (Fig. 7), a broadening of the contributions to the ν{FeIIC} vibration band is evidenced. This broadening reveals a perturbation of the Fe(CN)6 units due to the presence of the large cesium cations, with respect to smaller ones (as rubidium cations; Fig. 9).
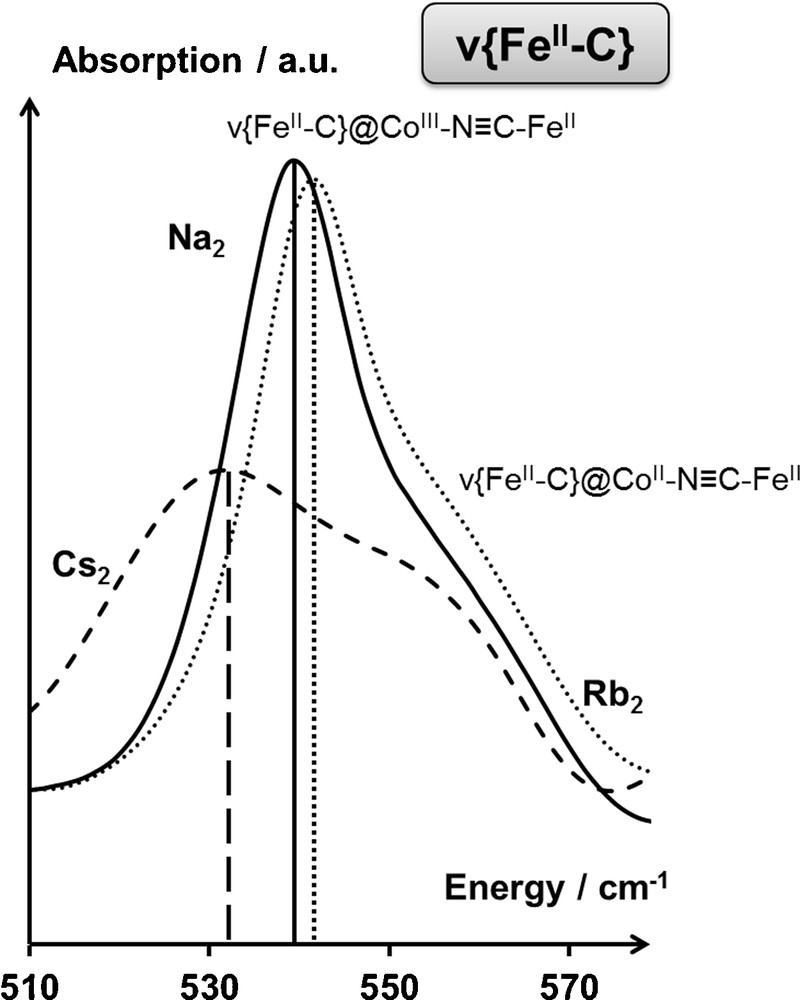
Infrared spectrum of the Na2CoFe (—), Rb2CoFe () and Cs2CoFe (— — —) Prussian blue analogues in the CoIIIFeII state (T = 200 K) in the ν{FeIIC}. Assignments are detailed in the main text.
This last series of examples highlights the suitability of IR spectroscopy to the fine investigation of the weak interactions between the alkali cation and the bimetallic network in PBAs, hardly detectable at the macroscopic scale, taking place in PBAs.
4 Conclusion
The investigation by IR spectroscopy of switchable PBAs allowed the comprehensive assignment of the IR spectrum of PBAs over the whole 150–4000 cm−1 spectral range, including vibration bands associated with metal–ligand bonds, cyanide bridges and water molecules that are not involved in a network of hydrogen bonds. The existence of minority species, such as CoIIN≡CFeII linkages in AxCoFe PBAs, has been pointed out. Direct evidences of the interaction between the inserted alkali cations and the bimetallic network taking place in PBAs have been demonstrated, paving the way to the study of the position of the alkali cation within the lattice of PBAs. This work constitutes a milestone in the study of PBAs and related molecular compounds by providing a reliable reference for the comprehensive assignment of the IR spectrum of such systems.