

1 Introduction
Decatungstate anion W10O324− is among the most photochemically active polyoxometalates and promotes transformations of organic substrates from alkanes to complex materials [1–11]. Illumination of W10O324− leads to the formation of a locally excited oxygen-to-metal charge-transfer state W10O324−*. This state decays in less than 30 ps to an extremely reactive and non-emissive transient [2] designated as wO [3]. This latter intermediate, which in the case of sodium decatungstate has a lifetime τwO of 65 ± 5 ns in acetonitrile, is the reactive species in photocatalytic systems. In the presence of a so-called XH substrate, wO reacts to produce a derived radical, X•, and the one-electron-reduced form (RF or H+W10O325−) W10O325− and/or protonated form.
Both RF and X• react with molecular oxygen to regenerate the active form (W10O324−) of the catalyst and form peroxy compounds [3,6,8–10]. Moreover, no chain reactions are observed [6].
It has recently been shown that acetone as well as acetonitrile and water can be considered a conventional solvent for polyoxometalates and especially decatungstate photocatalyzed reactions [12]. By comparison, it was concluded that the reactivity of wO with methanol is so high that the reactive intermediate is quantitatively scavenged by the solvent during the laser pulse. Hence, the nanosecond time scale laser flash photolysis technique is not adapted for investigations in alcohols. One might also wonder the effects on the photocatalytic cycle and the kinetic parameters in a system where methanol is a substrate in acetonitrile solution to one in which it acts as the solvent and, at the same time, as the substrate. Here, we will mainly focus on the decatungstate photocatalyzed oxygenation of dilute solutions of methanol in acetonitrile. The investigation on methanol as a solvent and a reactive substrate will be described elsewhere for clarity reasons.
Primary and secondary saturated alcohols, being selectively oxidized to the corresponding aldehydes and ketones, are widely used as substrates in mechanistic studies in polyoxometalates photocatalysis. For decatungstate, it has been established that the reaction of wO with aliphatic alcohols involves hydrogen-atom abstraction [1,3,7,13]. Based on the combination of time-resolved (laser flash photolysis) and steady state (oxygen consumption measurements) techniques, kinetic isotope effects, Hammett kinetics and product analysis, it has recently been demonstrated that the same conclusion applies for aromatic alcohols [14]. However, despite the fact that the photochemistry reaction is fairly well known, some aspects, especially the ones regarding the kinetics, deserve to be further assessed.
In the present work, it will be shown that:
- • a combination of steady state and time-resolved techniques provides access to the most important chemical and photophysical kinetic parameters of the reaction;
- • the reactivity of methanol is significantly lower than other already studied alcohols;
- • decatungstate photocatalyzed oxygenation concentration up to 2.5 M of methanol in acetonitrile satisfies the photostationary state conditions;
- • peroxyl radical (and O2) reoxidizes H+W10O325− to regenerate the catalyst and thus close the catalytic cycle;
- • progressive establishment of anaerobic conditions results in both ending of the substrate conversion and the decay of hydrogen peroxide.
2 Experimental
2.1 Materials
Sodium decatungstate was synthesized and purified following literature procedures [2a]. All other chemicals were purchased at the highest purity available from Aldrich, and were used as received. All experiments described in this work have been performed in acetonitrile, methanol or a mixture of both.
2.2 Laser flash photolysis
Laser flash photolysis studies were made with the third harmonic of a Q-switched Nd:YAG laser, at 355 nm. Solutions of sodium decatungstate were made up to 1.1 × 10−4 M, corresponding to an absorbance value of ∼ 0.4 at 355 nm. The sample solution, placed in a quartz cell having 1 cm path length, was excited with single laser pulses (100 mJ) and analyzed with a pulsed Xe arc lamp. The sample solution was refilled after each excitation using a flow system. Spectra were compiled point-by-point and kinetic studies were made at fixed wavelength. The reactive transient wO lifetimes were obtained by computational extrapolation of the first order decay profiles recorded at 780 nm. The RF formation quantum yields ΦRF were obtained from the 450 ns absorption after the pulse when the decay of wO at 780 nm is over, using the wO transient as an internal actinometric standard at this wavelength, according to the equation:
2.3 The photostationary state method
The photostationary state method consists in measuring the rate rOX of substrate photooxygenation by following oxygen consumption as a function of irradiation time. The corresponding quantum yield ΦOX is defined according to:
These experiments followed the general procedures described previously for the measurement of the quantum yield of singlet oxygen production under carefully controlled conditions [15]. Briefly, all photoreactions were carried out in an internal cylindrical photoreactor (volume 100 mL) illuminated with a Philips HPK 125 mercury lamp. The apparatus consists of a closed system including a solution of decatungstate (classically 5 × 10−4 M, corresponding to an absorbance value of ∼ 0.4 at 355 nm) in acetonitrile, or in methanol, or in a mixture of acetonitrile and methanol. 250 mL of O2 were also incorporated into the system. A vigorous O2 gas pumped stream provides a rapid circulation of the solution and supplies simultaneously the right amount of dissolved O2 that will be consumed for the reaction. The consumption of oxygen was measured under steady state irradiation using a gas buret. O2 consumption profiles generally display a short equilibration period, which is due to lamp stabilization and related to thermal effects resulting from the lamp heating. Light intensities were calibrated using the photooxygenation of furfuryl alcohol in O2 saturated acetonitrile with phenalenone (ΦOX = 0.98 [16]) as sensitizer. The concentration of decatungstate was sufficient to absorb the whole of incident light. The intensity of absorbed light was 6.97 × 10−6 einstein.s−1. Peroxide concentrations were determined by iodometric titration [17].
3 Results and discussion
3.1 Steady state photolysis
Previous studies have shown that oxygen consumption measurements represent a very useful tool to investigate the reactivity of wO towards organic substrates [3,6,8–10]. Fig. 1 shows the profiles of O2 consumption upon irradiation of oxygen-saturated acetonitrile solutions of sodium decatungstate, containing various concentrations of methanol. After a short equilibration period due to the lamp stabilization, consumption of O2 increases linearly with irradiation time. The relative rates of O2 consumption are calculated from the slope of the linear portion of these plots. They can be converted into quantum yield values by comparison with the rate of O2 consumption for phenalenone-sensitized oxygenation of furfurylic alcohol (quantum yield 0.98 [16]). Fig. 2 shows that the O2 consumption quantum yield, ΦOXXH, increases progressively with increasing concentration of substrate up to 1.0 M, and that the value in the absence of any substrate ΦOX remains finite. Thus confirming that acetonitrile itself acts as a substrate for wO [3].

Time dependence of O2 consumption measured for illumination of oxygen-saturated solutions of sodium decatungstate (5.5 × 10−4 M) in acetonitrile in the absence (□) and presence of methanol at 0.1 M (•), 0.2 M (Δ), 0.3 M () and 0.4 M (ο).
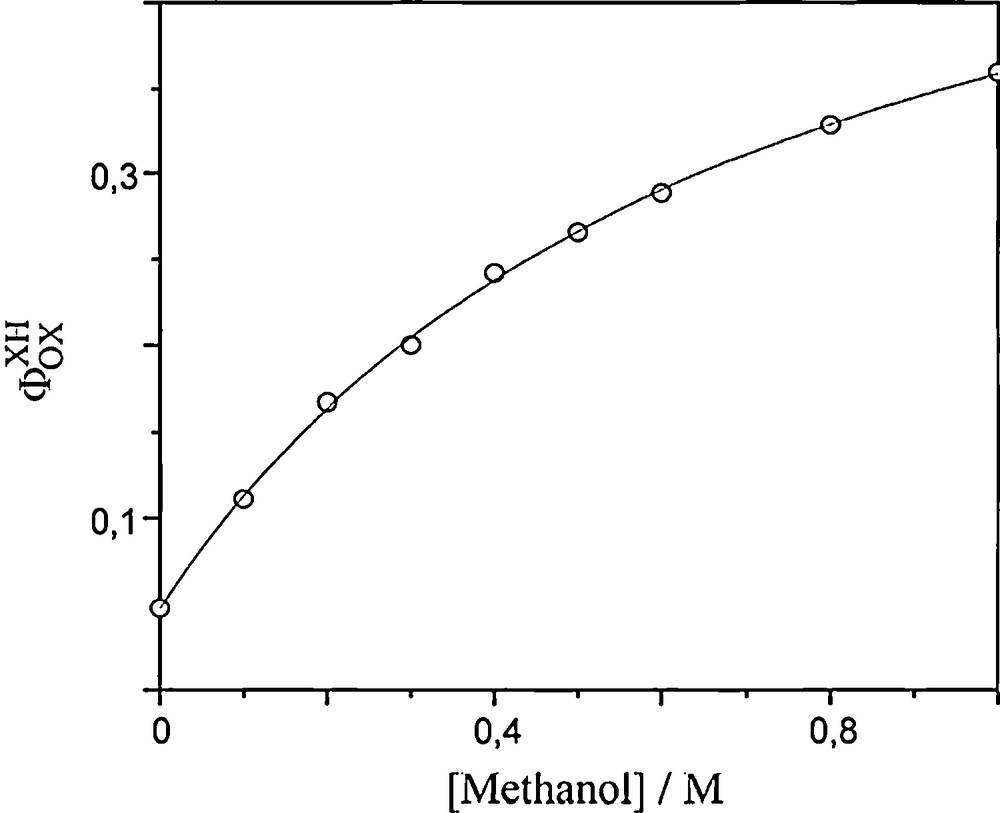
Effect of methanol concentration on the quantum yield of oxygen consumption measured for 5.5 × 10−4 M sodium decatungstate in oxygen-saturated acetonitrile. The solid line is drawn in accordance with equation ФXHOX = (ФOX + ФwOK[XH])/(1 + [XH]), with ФOX = 0.048, ФwO = 0.58, and K = 1.37 M−1.
These data can be used to derive the rate constant kXH for the reaction between wO and methanol by considering the process in terms of a generalized Stern-Volmer treatment [3,6,10]:
Transient absorbance at 780 nm and oxygen consumption as a function of substrate.
| Transient absorbance at 780 nm | Oxygen consumption | |||||||
| wO decay | W10O325−formation | |||||||
| Substrate | kXH/107 M−1s−1 | K/M−1 | ФRF0 | ФRF∞ | K/M−1 | ФOX | ФwO | K/M−1 |
| Benzyl alcohol [14a] | 28 | 19.4 | 0.023 | 0.45 | 19.1 | 0.047 | 0.56 | 20.1 |
| 1-phenylethanol [14a] | 14.4 | 9.9 | 0.024 | 0.47 | 8.1 | 0.047 | 0.56 | 9.9 |
| Propan-2-ol [3,12] | 9.8 | 5.8 | 0.022 | 0.33 | 4.8 | 0.048 | 0.57 | 5.6 |
| Acetone [12] | 4.9 | 3.1 | 3.2 | |||||
| Methanol | 1.9 | 1.3 | 0.023 | 0.36 | 1.46 | 0.048 | 0.58 | 1.37 |
Illumination in the presence of O2 results in the formation of hydrogen peroxide derived from the substrate and O2. It has been shown that in most of the cases, decatungstate catalyses conversion of propan-2-ol into acetone and hydrogen peroxide in quantitative yield [3]. Fig. 3 shows the time profile of the oxygen consumption and peroxide formation during decatungstate catalyzed photooxygenation of 0.8 M methanol in air and oxygen-saturated acetonitrile in the case of a closed reactor. The initial oxygen concentration is fixed and the concentration of dissolved molecular O2 decreases progressively as its reaction with the substrate proceeds until anaerobic conditions are reached. This approach has been used in order to evaluate the impact of a change in the concentration of the other reactant, the molecular oxygen, which may occur if the rate rOX of the substrate photooxygenation is large enough to result in oxygen depletion in the medium. It has been observed that:
- • the initial variations for the first 20–25 minutes or so are linear and that the rate of peroxide formation is equal to the rate of the O2 consumption independently of the concentration of oxygen (O2 or air);
- • the decay of hydrogen peroxide starts somewhere before the end of the O2 consumption and thus before anaerobic conditions are completely established.
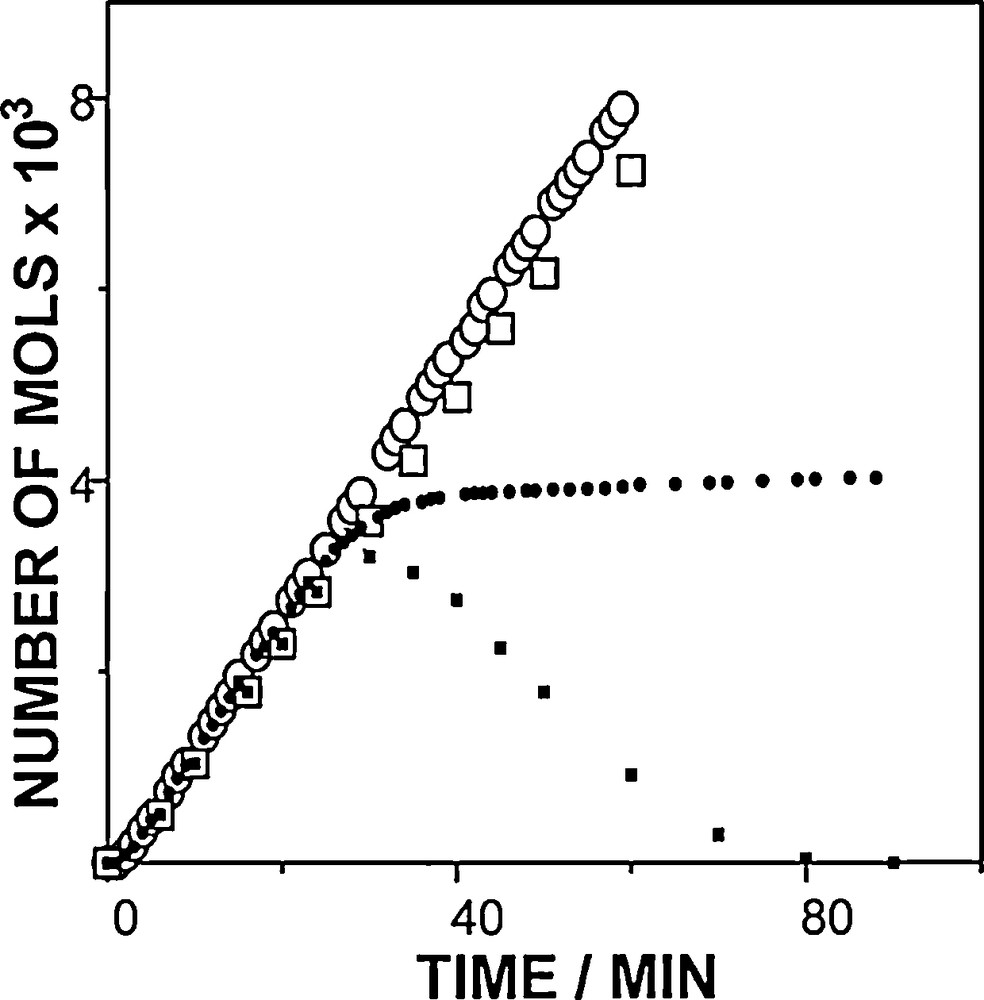
Correspondence between the number of mols of oxygen consumed (circle) and of peroxide formed (square) during continuous illumination of sodium decatungstate (5.5 × 10−4 M) in oxygen (open symbols) and air (solid symbols) saturated acetonitrile in the presence of methanol at 0.8 M.
For O2 consumption, a plateau value is observed around 40 minutes for air-saturated solutions, and for peroxide, a concentration maximum is observed around 30 minutes. This clearly confirms that the change in O2 consumption kinetics is related to the establishment of anaerobic conditions that have not yet been reached for O2 saturated solutions. It also establishes that wO reacts with the reaction product H2O2 which is no longer formed when molecular oxygen, its source, is removed from the medium.
The reoxidation of the reduced form of decatungstate by peroxyl radicals that may be considered as a termination reaction in the overall radical transformation [6] is an important process for the regeneration of the catalyst. However, under continuous photolysis conditions, in a well-agitated system of oxygen-saturated acetonitrile or methanol under low near-UV irradiance, the concentration of O2 is much higher than the steady state concentration of wO and any radical. Therefore, direct reoxidation of H+W10O325− by O2 may play a major part in the regeneration of the catalyst even if the rate constant of the reaction between the reduced form with oxygen is low [4,18].
For concentrations below 2.5 M of methanol in acetonitrile, the mechanism of the decatungstate photocatalysis may be schematized by the catalytic cycle shown on Eq. (1).
| (1) |
The transient wO reacts with methanol to give the one-electron-reduced species H+W10O325− and the corresponding hydroxymethyl radical. This latter is quantitatively intercepted by oxygen and reacts at a near diffusion-controlled rate to form the hydroxymethylperoxyl radical. Then, the peroxyl radical reoxidizes H+W10O325− to regenerate the catalyst and gives the reaction products, thus closing the catalytic cycle. More generally, it is interesting to note that Eq. (1) is slightly oversimplified since oxygen not only intercepts the freely diffusing •CH2OH radicals but also the geminate ion pairs [H+W10O325−,•CH2OH], as mainly argued about alkenes [10].
Fig. 2 shows that the relationship ФXHOX = (ФOX + ФwOK[XH])/(1 + [XH]) directly derived from equation (A) fits quite perfectly the variations of ФXHOX vs. [XH] for concentrations up to 1 M in acetonitrile. Actually, it is the same until [CH3OH] reaches ∼ 2.5 M from which point the quantum yield ФXHOX slows down very surprisingly after a maximum value of ∼ 0.45. It even decreases from 5 M to pure methanol (data not shown). This bend is observed for both oxygen- and air-saturated acetonitrile-methanol mixtures. The yields always remain lower in the presence of air. Relation (A) is no longer satisfied showing that photostationary state conditions are no longer fulfilled meaning:
- • that the formation and disappearance rates are not equal for each of the transient entities formed;
- • that in the present case, the steady state approximation does not apply to the reduced form of the catalyst;
- • and therefore that the rate of reoxidation of H+W10O325− became too low in comparison with the rate of photooxygenation rOX.
For similar experiments in which acetone acts as a substrate in acetonitrile, the quantum yield ФXHOX increases progressively up to a value of 0.175 in pure CH3COCH3 but no curve was observed [12]. This difference can now be explained by the fact that the rate of photooxygenation remains always much less than that observed with methanol. This system meets the photostationary state conditions regardless of the acetone concentration.
3.2 Laser flash photolysis
Fig. 4 shows kinetic traces recorded at 780 nm following a 355 nm excitation of air saturated acetonitrile solutions of decatungstate (5 × 10−4 M) in the presence of variable concentrations of methanol. These decays correspond to the wO transient and the related pseudo-first-order rate constants have been plotted in the corresponding Stern–Volmer graph (Fig. 5). The so-determined slope yields kXH = 1.88 × 107 M−1 s−1 (Table 1). The absorbance baseline is not reformed after wO decay and it has been previously shown that this absorbance is due to the one-electron-reduced form of decatungstate, W10O325−, or its protonated form HW10O324−. The transient absorbance change recorded at 780 nm, 450 ns following laser excitation, may be used to calculate the quantum yields ФRFXH of RF formation (see Section 2) and may be expressed in terms of a generalized Stern–Volmer treatment [10]:
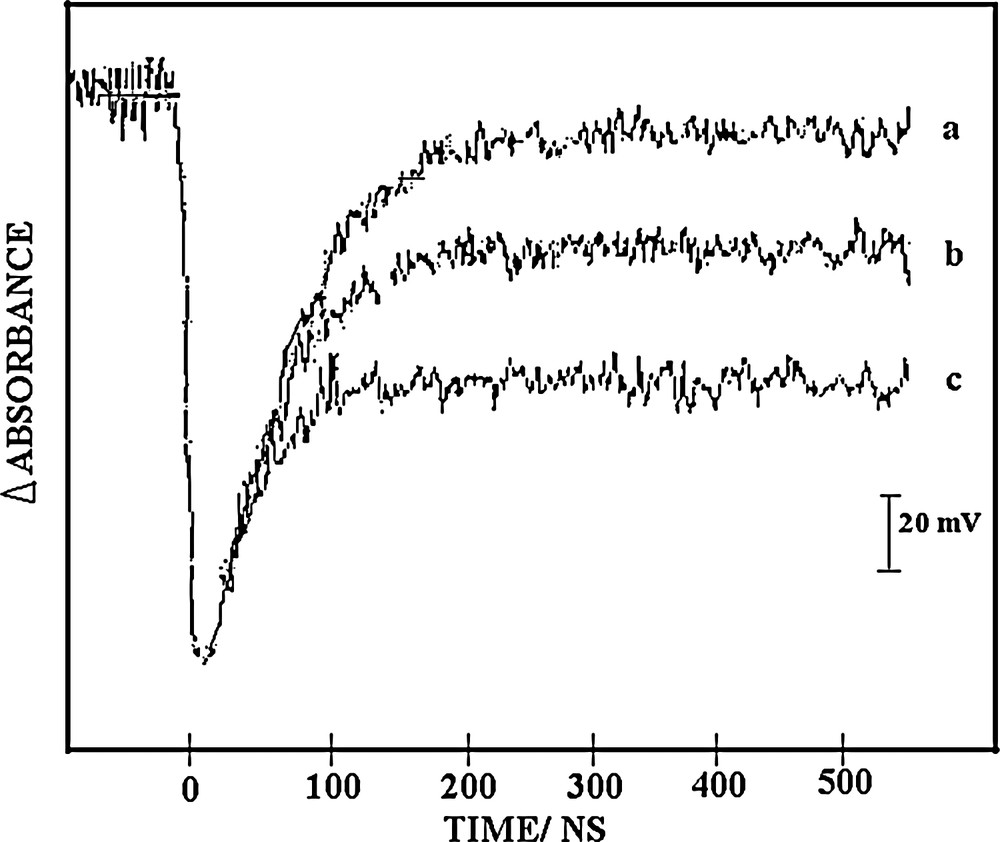
Kinetic profiles observed at 780 nm showing decay of wO as generated following 355 nm excitation with a 15 ns laser pulse of 5 × 10−4 M sodium decatungstate air-saturated acetonitrile solution in the absence (a) and presence of methanol at 0.25 M (b) and 1.0 M (c).
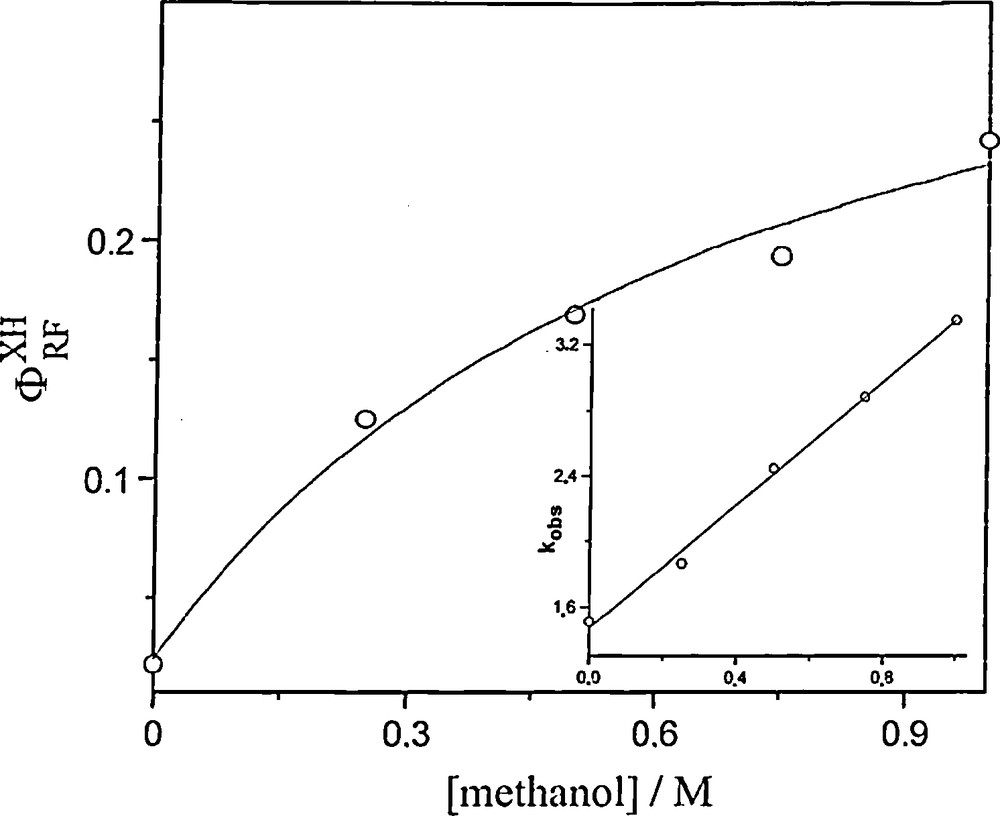
Dependence of the quantum yield for formation of the reduced form of decatungstate on methanol concentration, following the 355 nm pulsed excitation (∼ 20 mJ) of air-saturated acetonitrile solutions of sodium decatungstate (5.5 × 10−4 M). The solid line is drawn is accordance with the equation ФXHRF = (ФRF0 + ФRF∞K[XH])/(1 + K[XH]), with ФRF0 = 0.023, ФRF∞ = 0.36 and K = 1.46 M−1. Inset: dependence of the corresponding pseudo-first-order rate constant of decay of wO measured at 780 nm on methanol concentration in air-saturated acetonitrile.
The spectrum of the long-lived species generated by a 355 nm excitation with a 15 ns laser pulse of a 5 × 10−4 M sodium decatungstate air-saturated acetonitrile solution in the presence of methanol at 4.5 M is given on Fig. 6. This spectrum is the same as the one obtained under identical conditions by excitation of a tetrabutylammonium decatungstate air-saturated propanol-2-ol/acetonitrile solution and also as the one resulting from an injection of a 20 ns pulse of high-energy electrons into a tetrabutylammonium decatungstate N2-saturated solution after 220 μs [3]. Thus, the long-lived species of Fig. 6 can well be identified as the one-electron-reduced species.
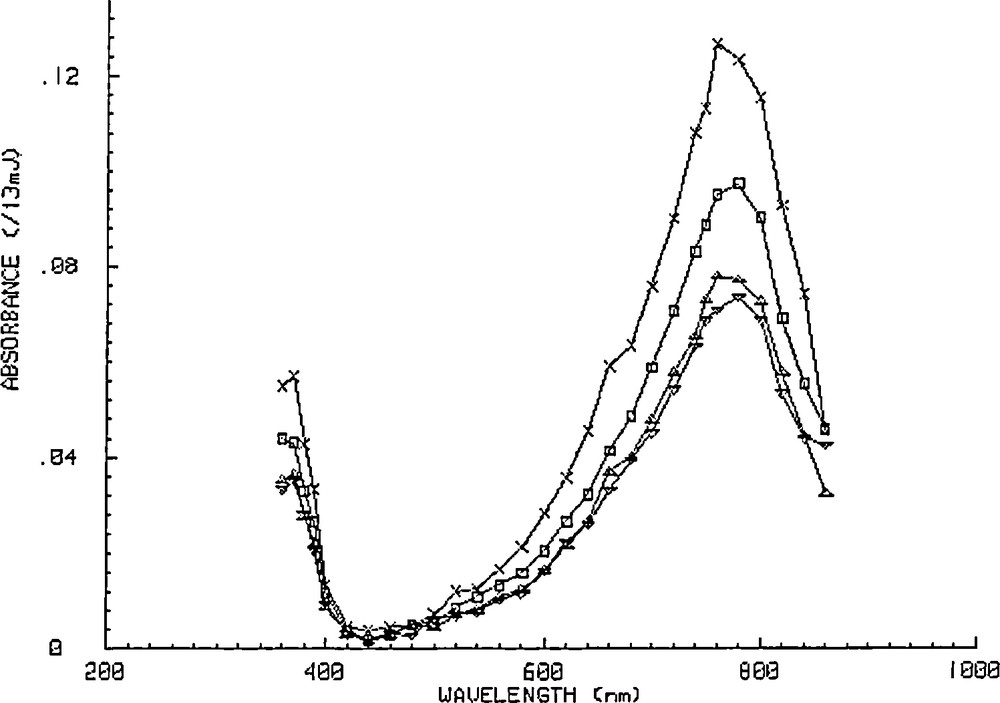
Transient absorption spectrum recorded for the one-electron reduced form generated by 355 nm excitation with a 15 ns laser pulse of a 5 × 10−4 M sodium decatungstate air-saturated acetonitrile solution in the presence of methanol at 4.5 M after 30 μs (×), 320 μ (□), 1.53 ms (Δ), and 4,27 ms (▿).
For comparison purpose, the kinetic parameters obtained for methanol, but also for other alcohols and acetone are collected in Table 1. It is noteworthy:
- • that the three values of Stern–Volmer constants, obtained independently from both steady state and time-resolved measurements are almost identical for each of the substrates;
- • that the same is true for the values of the kinetic parameters that characterize decatungstate photocatalysis in acetonitrile such as ФwO, ФOX, and ФRF0.
This indicates that all approaches provide reliable data and give a good overview of the experimental uncertainties associated with the individual measurements.
Previous studies involving a wide variety of organic substrates suggested that the quenching of wO may occur by hydrogen-atom-abstraction and/or electron transfer mechanisms depending on the chemical nature of XH [3–14]. In all cases, both mechanisms lead to the same one-electron-reduced species and to the corresponding substrate-derived radical. With alkanes and aliphatic alcohols, the quenching of wO occurs by hydrogen-atom-abstraction, whereas with any easily oxidizable substrates, such as aromatic hydrocarbons [11] and alkenes [10], direct electron transfer can compete with hydrogen atom transfer and even become the main pathway.
As aromatic alcohols are also potentially capable of reacting following both mechanisms, a combination of steady state and time-resolved techniques with kinetic isotope effects studies has recently been used to investigate the mechanism of their decatungstate photocatalyzed oxidation [14]. It has been demonstrated that they exclusively react by hydrogen atom transfers [14a]. In addition, the primary and secondary isotope effects provided strong evidences of a stepwise mechanism where the hydrogen-atom-abstraction occurs in the rate-determining step of the reaction. Finally, to examine further the extent of the electronic effect on the reaction rates and to obtain valuable information on the transition state TS of the decatungstate photocatalyzed reaction of 1-phenylethanol, the Hammett kinetics of the oxidation on a series of para-substituted-1-phenylethanols has been studied [14b]. A small positive slope has been found that:
- • it indicates the development of negative charge or a radical intermediate which is better stabilized by an electron-withdrawing substituent in the transition state and that;
- • it suggests a radical intermediate for this reaction and supports an early transition state.
Compared to aliphatic alcohols such as methanol whose intermediate in the TS is the hydroxymethylperoxyl radical [Eq. (1)], the stabilization of the transition state for aromatic alcohols results in a decrease of the activation energy and then, in an increase of the rate constant kXH which explains the significant higher reactivity of the latter (Table 1).
For aliphatic alcohols, the rate constant of wO with methanol is much less than with secondary compounds such as propan-2-ol which reflects that the process involves in this case primary CH bonds and not secondary CH bonds as seen for propan-2-ol. The higher reactivity of acetone is due to the fact that it is a solvent/substrate bearing 6 activated primary CH bonds (Table 1). This result relative to the simplest alcohol completes the study made on the reactivity of many different linear and branched aliphatic alcohols [3], and confirms the hydrogen-atom-abstraction mechanism.
4 Conclusions
In decatungstate photocatalyzed oxygenations, two main parameters control the reactivity of the reactive species wO with alcohols. The first one is the CH binding energy, as expected on the basis of a hydrogen-atom-abstraction mechanism. Therefore, the bimolecular rate constant of methanol with the reactive oxyradical wO is the lowest ever observed for aliphatic alcohols. The second determining parameter is the nature of the alcohol that governs, in the transition state, the electronic effects and thus its energy level and its reactivity. Due to a better stabilization of the radical cation between XH and wO that is developing in the transition state, the rate constant kXH of aromatic alcohols is significantly higher than that of aliphatic alcohols (Table 1).
In addition to the study of the elementary reaction between wO and XH, two new and unpublished experimental observations in the photocatalysis by decatungstate should be highlighted. First of all, for methanol concentrations above 2.5 M the quantum yield of photooxygenation no longer satisfies the kinetic relationships deduced from the steady state approximation and the system no longer obeys the photostationary state conditions. Secondly, progressive establishment of anaerobic conditions, which may occur if the rate of oxygenation becomes higher than that of catalyst regeneration, results in both ending of substrate conversion and the decay of hydrogen peroxide. These findings open up perspectives on the development of a new methodology for studying the parameters and processes that control the nature of the slow step and the progress of the photocatalytic cycle. This methodology is currently under investigation and is applied to the study of decatungstate photocatalyzed oxygenation of methanol and higher aliphatic alcohols as solvent and reactive substrate.
Acknowledgments
The laser flash photolysis studies were performed at the Paterson Institute for Cancer Research Free Radical Research Facility, Manchester, UK, with support of the European Commission through the Access to Large-Scale Facilities activity of the TMR Program. Dr. Ioana Fechete is gratefully acknowledged for her assistance in the preparation of figures and Murielle Oster for the improvement of English spelling.