

1 Introduction
Optically active amine scaffolds such as 2-aminotetraline derivatives are found in many biologically active molecules. This family of compounds provides very important intermediates for organic synthesis and members are present in drug candidates and pharmaceutical compounds (Fig. 1) [1]. Indeed, 2-aminotetraline derivatives have been reported to exhibit, for example, anti-inflammatory activity [2]. More, ST1214 [3] has been developed as a potent drug for cerebral ischemia and stroke. 7-OH-DPAT [4] was found to be a selective dopamine D3 receptor antagonist and CHF-1024 [5] is a compound endowed with DA2-dopaminergic/α2-adrenergic receptor antagonist activity in myocardial ischemia/reperfusion damage. Terutroban [6] is a thromboxane/prostaglandin endoperoxide receptor antagonist. The racemic sodium salt of terutroban (S18204) was described first in 1995 [7]. The synthesis includes a Diels–Alder reaction between the appropriate 2-pyrone and a properly substituted alkyne (Scheme 1) [8]. The optically pure (R) sodium salt of terutroban (S18886), which is the biologically active enantiomer, was obtained by chiral HPLC separation of enantiomers [8a].
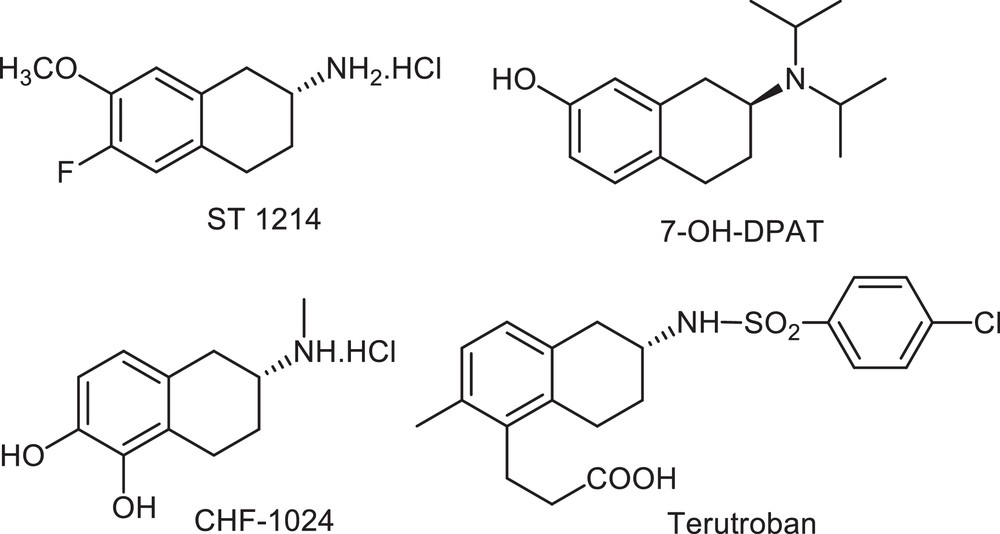
Examples of chiral 2-aminotetralines based pharmaceutical compounds.
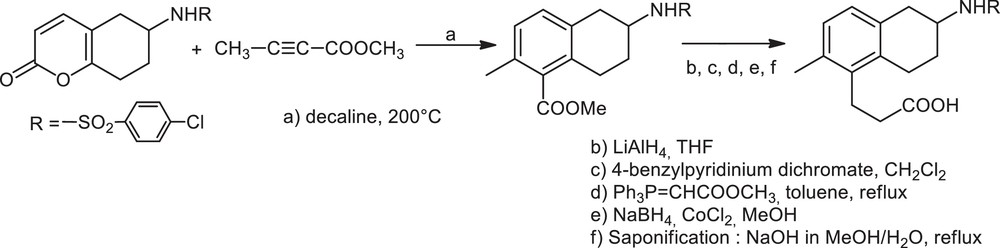
Synthesis of racemic terutroban.
Synthesis of optically pure 2-aminotetralines via asymmetric hydrogenation is a powerful and straightforward methodology. Different precursors to optically enriched 2-aminotetralines can be used such as imines [9], iminiums [10], enamines [11], enecarbamates [12], and enamides [13], in the presence of a large variety of catalysts based mainly on chiral organometallic complexes of rhodium, ruthenium, and iridium.
In order to obtain the optically enriched 2-aminotetraline scaffold of terutroban, direct asymmetric hydrogenation of a key intermediate is the most direct method. An enecarbamate precursor has been hydrogenated for that purpose as reported by Bruneau and Dixneuf (substrate/ruthenium catalyst = 100, 70% yield, 92% ee) [12]. In spite of this interesting result, the methodology suffers from low substrate/catalyst ratio, which limits its potential application.
As part of our ongoing research toward the use of catalyzed asymmetric transformations for the synthesis of biologically relevant targets [14], previously, we have also reported on the enantioselective access to chiral 2-aminotetraline derivatives and disclosed a highly selective synthesis of Amibregeon (SR58611A), a potent selective β3 adrenergic receptor agonist [15]. We thought to explore further the asymmetric hydrogenation of enamides to prepare a (R) optically enriched intermediate of terutroban.
2 Results and discussion
The enamide 3-(6-acetamido-2-methyl-7,8-dihydronaphthalen-1-yl)propanoic acid 1 has been selected as substrate (Scheme 2). The latter was synthesized following a classical procedure by the condensation of acetamide with the corresponding cyclic ketone in the presence of para-toluenesulfonic acid as catalyst [12a,13b]. Then, in a typical hydrogenation experiment, either a commercially available ruthenium precatalyst was used or the catalyst precursor was prepared easily by mixing a ruthenium complex and the desired chiral phosphorous ligand in 15 mL of methanol under an inert atmosphere. In the latter case, the mixture was stirred at room temperature for 1 h before transfer into a double walled 50 mL stainless steel autoclave, which contained already substrate 1. Then, the autoclave was pressurized with hydrogen and heated to the desired temperature. For all catalytic assays, methanol was used as solvent for a better solubility of the substrate (Table 1). As a consequence, methyl esters could be formed in the catalytic medium. Indeed, beside the expected hydrogenation product 2, we identified the methyl ester of the hydrogenation product 3 and the esterified substrate 4. In order to simplify analyses, the crude reaction mixtures were always reacted with 0.3 mL of NaOH 10 N at 40 °C for 4 h. Doing so, only the remaining substrate 1 and the hydrogenated product 2 were present and the mixtures could be analyzed easily by HPLC to determine yields and selectivities.

Hydrogenation of the 3-(6-acetamido-2-methyl-7,8-dihydronaphthalen-1-yl)-propanoic acid 1.
Asymmetric hydrogenation of the enamide 1 in the presence of commercially available or in situ generated ruthenium catalystsa.
| Entry | Ru precursor | Conv. (%)b | Yield 2 (%)b | Ee 2 (%)b |
| 1 | Ru(OAc)2(S)-Binap | 98 | 92 | 84 (R) |
| 2c | Ru(OAc)2(S)-Binap | 94 | 89 | 86 (R) |
| 3 | Ru(OAc)2(S)-Tol-Binap | 98 | 72 | 82 (R) |
| 4 | Ru(OAc)2(S)-H8-Binap | 97 | 88 | 73 (R) |
| 5 | Ru(OAc)2(S)-Segphos | 97 | 80 | 82 (R) |
| 6 | Ru(OAc)2(S)-Tol-Segphos | 96 | 71 | 82 (R) |
| 7 | Ru(cod)(methylallyl)2 + (S)-Segphos | 100 | 93 | 86 (R) |
| 8 | Ru(cod)(methylallyl)2 + (S)-MeO-Biphep | 100 | 86 | 86 (R) |
| 9d | Ru(cod)(methylallyl)2 + (S)-MeO-Biphep | 100 | 100 | 85 (R) |
| 10e | Ru(cod)(methylallyl)2 + (S)-MeO-Biphep | 100 | 93 | 85 (R) |
a S/Ru = 500; MeOH, 15 mL; P(H2) = 100 bar; T = 60 °C; t = 17 h.
b Determined by HPLC.
c P(H2) = 80 bar and T = 70 °C.
d S/Ru = 250; MeOH/acetone: 2/1, 15 mL; P(H2) = 80 bar; T = 70 °C; t = 17 h.
e S/Ru = 2000; MeOH/acetone: 2/1, 15 mL; P(H2) = 80 bar; T = 70 °C, t = 21 h.
For our first series of experiments, we studied commercially available ruthenium diacetate catalytic precursors bearing atropisomeric diphosphine ligands i.e. (S)-Binap, (S)-Tol-Binap, (S)-H8-Binap, (S)-Segphos, and (S)-Tol-Segphos (Fig. 2). The reactions were carried out under 100 bar H2 and at 60 °C for 17 h with a substrate/catalyst ratio as high as 500. The results gathered in Table 1.

Ligands of commercially available Ru-catalysts and (S)-MeO-Biphep ligand.
In several cases, the conversions were high (94–98%) (entries 1, 3, 4–6), but the desired hydrogenated product was accompanied by unidentified compounds (71–92% yield, 84% ee). By-products have not been isolated. Nevertheless, thanks to LC–MS analyses, we can propose a product with a free amine and one corresponding to the primary alcohol issued from the reduction of the acid moiety of the hydrogenated product 2.
We varied the hydrogen pressure and the reaction temperature and found that the optimal reaction conditions in terms of activity and enantioselectivity were 70 °C and 80 bar H2. For example, results obtained in the presence of Ru(OAc)2(S)-Binap under 80 bar of hydrogen and at 70 °C (89% yield, 86% ee) (Table 1, entry 2) are very close to those acquired at 100 bar and at 60 °C (92% yield, 84% ee) (Table 1, entry 1). Next, we examined catalysts generated in situ by mixing Ru(cod)(methylallyl)2 and bisphosphine ligands (entries 7–10) [16]. Interestingly, in the case of (S)-Segphos, we obtained better results in terms of activity (93% yield) and enantioselectivity (86% ee) (Table 1, entry 7 vs. 5). The catalytic system based on (S)-MeO-Biphep (Fig. 2) under identical catalytic conditions provided also a high enantioselectivity of 86% (entry 8). We observed that the use of the mixture acetone/MeOH as solvent allowed reducing the formation of by-products (Table 1, entry 9 vs. 8) and led to 100% yield with a similar enantioselectivity (85% ee). In addition, we performed a catalytic test with a substrate/Ru ratio of 2000. The hydrogenation was almost complete within 21 h and we determined a yield of 93% and an enantioselectivity of 85% (Table 1, entry 10). The desired product was isolated through silica gel chromatography and the chemical yield and enantiomeric excess are consistent with the data displayed in Table 1 (entry 10).
In order to improve further the enantioselectivity, other chiral ligands have then been examined (Fig. 3). Catalytic reactions were carried out in a mixture of MeOH and acetone 2/1 (15 mL) under the mildest conditions (80 bar, 70 °C). The catalysts were generated in situ from Ru(cod)(methylallyl)2, the substrate/catalyst ratio being 250.
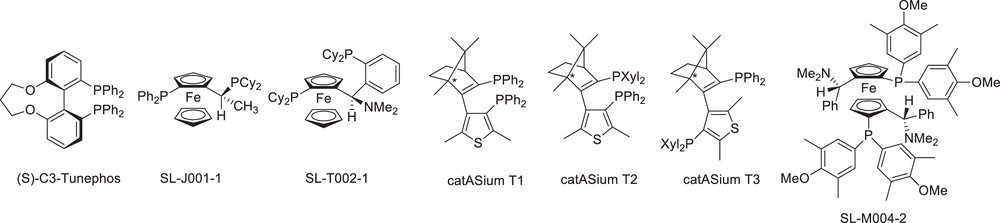
Bisphosphine ligands applied in asymmetric hydrogenation of eneacetamide 1.
The results gathered in Table 2 show that the enantioselectivities were strongly dependent upon the nature of the ligand. Interesting features appeared. First, the ligands with an atropisomeric skeleton as (S)-Binap, (S)-MeO-Biphep and (S)-C3-Tunephos (entries 1–3) led to ee's close to 84%. The use of catASium®T ligands was more interesting (entries 7–9). This ligand family combines a central chirality derived from a natural product with axial chirality [17]. Using catASium®T ligands, up to 92% ee were obtained. At a 250 substrate/Ru ratio, the reaction was complete within 17 h. We increased the ratio up to 500 with the Ru/catASium®T3 system, and after 17 h, 100% conversion was still obtained with 89% yield and 90% enantioselectivity. A higher ratio of 1000 allowed always a complete conversion after the same reaction time with 90% yield but we observed an erosion of the ee to 68%. It has to be noticed that in that case, the optically pure amide 2 precipitated in the mixture as a grey–white solid (60% yield).
Asymmetric hydrogenation of the enamide 1 in the presence of Ru(cod)(methylallyl)2 and a diphosphinea.
| Entry | Ligand | Conv. (%)b | Yield 2 (%)b | Ee 2 (%)b |
| 1c | (S)-Binap | 94 | 83 | 84 (R) |
| 2 | (S)-MeO-Biphep | 100 | 100 | 82 (R) |
| 3 | (S)-C3-Tunephos | 100 | 92 | 86 (R) |
| 4 | J001-1 | 97 | 95 | 0 |
| 5 | T002-1 | 96 | 93 | 49 (S) |
| 6 | M004-2 | 50 | 45 | 11 (S) |
| 7 | (1S,4R)-catASium®T1 | 96 | 90 | 88 (R) |
| 8 | (1S,4R)-catASium®T2 | 98 | 96 | 92 (R) |
| 9 | (1R,4S)-catASium®T3 | 100 | 97 | 90 (S) |
a S/Ru = 250; MeOH/acetone: 2/1, 15 mL; P(H2) = 80 bar; T = 70 °C; t = 17 h.
b Determined by HPLC.
c t = 72 h, time not optimized.
In order to avoid the saponification step at the end of the hydrogenation reaction, other solvents as acetone, THF and CH2Cl2 have been applied. Results are reported in Table 3 using the chiral catASium®T3 ligand. In the case of THF, the ee was low (66%, entry 3). In dichloromethane, the conversion remained low, due to a low solubility of the substrate. Acetone was interesting in terms of activity and enantioselectivity but, as previously, unidentified products were formed (compare entries 1 and 2, conv. 100%, selectivity 86% and 97%, respectively). In pure acetone, we also observed a slightly lower 86% ee in comparison to the 90% value obtained in the mixture MeOH/Acetone. Nevertheless, in this latter case, saponification remained necessary.
Effect of solvent in the asymmetric hydrogenation of the ene acetamide 1 in the presence of Ru(cod)(methylallyl)2 and (1R,4S)-catASium®T3a.
| Entry | Solvent | Conv. (%)b | Yield 2 (%)b | Ee 2 (%)b |
| 1 | Acetone | 100 | 86 | 86 (S) |
| 2c | MeOH/acetone | 100 | 97 | 90 (S) |
| 3 | THF | 100 | 96 | 66 (S) |
| 4 | CH2Cl2 | 35 | 27 | ND |
a S/Ru = 250; solvent: 15 mL; P(H2) = 80 bar; T = 70 °C; t = 17 h.
b Determined by HPLC.
c Ee measured after saponification.
This study allowed determining that two catalytic systems are able to provide a very good compromise in terms of yield and enantioselectivity for an efficient asymmetric hydrogenation of 1. The first one, based on the chiral ligand (1R,4S)-catASium®T3, is applicable to a substrate/catalyst ratio of 500 leading to 89% yield and 90% ee. The second one, based on (S)-MeO-Biphep, is allowed reaching full conversion, 93% yield, and 85% ee into the desired product while applying a substrate/catalyst ratio of 2000. After the hydrogenation reaction, deprotection of the amide could be performed in high yield and without racemization in the presence of oxalyl chloride followed by addition of propylene glycol affording the optically pure amine [18], which is then easily elaborated into optically pure terutroban.
3 Experimental part
3.1 Materials and methods
All reactions were carried out under an inert nitrogen atmosphere using standard Schlenk techniques. THF and CH2Cl2 were obtained from a solvent purification system MBraun SPS-800. Methanol was distilled over CaH2. Acetone is HPLC grade. All the solvents were degassed prior to use. Proton nuclear magnetic resonance (1H NMR) spectra were recorded using a Bruker AC 300 spectrometer (300 MHz). Chemical shifts are reported in delta (δ) units, part per million (ppm) downfield from tetramethylsilane (TMS) relative to the singlet at 7.26 ppm for deuterated chloroform. Coupling constants are reported in Hertz (Hz). The following abbreviations are used: s, singlet, d, doublet, t, triplet, m, multiplet. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded using a Bruker 300 (75 MHz). Chemical shifts are reported in delta (δ) units, part per million (ppm) relative to the center line of the triplet at 77.16 ppm for deuterated chloroform. 13C NMR analyses were run with broadband decoupling. The conversion of the substrate and hydrogenation yields were determined by HPLC–RP8 3.5 mm (15 cm × 4.6 mm) column from waters (H2O + 0.1 methane sulfonic acid/acetonitrile = 60/40, flow = 1 mL/min, 40 °C, at 210 nm). The enantiomeric excesses were determined by HPLC analysis using Chiracel OJ-RH column from Daicel (H2O at pH = 2.0 with H3PO4/ethanol = 65/35, flow = 0.8 mL/min, 40 °C, 210 nm).
3.2 Preparation of 3-(6-acetamido-2-methyl-7,8-dihydronaphthalen-1-yl)propanoic acid 1
3-(2-methyl-6-oxo-5,6,7,8-tetrahydronaphtalen-1-yl)propanoic acid (1.18 g, 4.3 mmol) and acetamide (0.64 g, 10.8 mmol) were heated in the presence of p-toluenesulfonic acid monohydrate (0.082 g, 0.43 mmol) in 50 mL toluene in a Dean–Stark apparatus under nitrogen atmospher for a night. After cooling, the organic phase was washed three times with water, then evaporated to dryness. The amide 1 was purified by flash chromatography on silica gel with a mixture of diethylether/pentane as solvent.
Yield: 75%; 1H NMR (300 MHz, DMSO) 1.98 (3H, s, CH3), 2.23 (3H, s, CH3), 2.33 (4H, m, CH2), 2.79 (4H, m, CH2), 6.71 (1H, d, CHar), 6.88 (1H, d, CHar), 7.02 (1H, s, CH), 9.27 (1H, s, NH), 12.21 (1H, s, OH); 13C NMR (75 MHz, DMSO) 19.91, 24.18, 24.43, 24.65, 26.71, 33.96, 109.59, 124.19, 128.45, 131.05, 133.39, 133.68, 136.00, 136.15, 169.00, 174.41.
3.3 General procedure for the catalytic asymmetric hydrogenation of 3-(6-acetamido-2-methyl-7,8-dihydronaphthalen-1-yl)propanoic acid 1 (S/Ru = 1000)
MeOH, acetone, THF, and CH2Cl2 were degassed by nitrogen bubbling. A 50 mL Schlenk tube equipped with a magnetic stirrer was charged with Ru(cod)(methylallyl)2 (1.7 mg; 5.3 × 10−3 mmol) and the selected chiral ligand (5.5 × 10−3 mmol). Then, the reactor was degassed (three vacuum/nitrogen cycles) and the degassed solvent or solvent mixture (15 mL) was added. The mixture was stirred at room temperature for 1 h before cannula transfer into a degassed (three vacuum/nitrogen cycles) double walled 50 mL stainless steel autoclave, which contained already substrate 1 (5.5 mmol). After vacuum degassing (three cycles vacuum/nitrogen) and three purges with hydrogen, the autoclave was pressurized with hydrogen and heated. After 17 h, the heating was stopped and after cooling, hydrogen was purged. The crude mixture was passed through a little silica column before HPLC analyzes.
1H NMR (300 MHz, DMSO) 1.60 (1H, m, CH–CHNH), 1.82 (3H, s, CH3–CO), 1.96 (1H, m, CH-CHNH), 2.23 (3H, s, H3C–Ph), 2.55–2.89 (8H, m, 4 CH2), 3.87 (1H, CH–NH, m), 6.80 (1H, d, CHar), 6.91 (1H, d, CHar), 7.89 (1H, d, NH), 12.23 (1H, s, OH); 13C NMR (75 MHz, DMSO) 19.10, 22.70, 24.18, 24.52, 28.88, 32.83, 35.65, 44.23, 127.04, 127.66, 132.60, 133.15, 133.53, 136.83, 168.62, 174.06.
4 Conclusion
The catalytic asymmetric hydrogenation of the eneamide terutroban precursor has been efficiently performed in the presence of in situ generated ruthenium catalyst bearing catASium®T chiral phosphorous ligands. High activities and enantioselectivities (92% ee) have been obtained with a substrate/Ru ratio 250. Moreover, high levels of activity and selectivity are maintained at ratios up to 2000 in the presence of a ruthenium catalyst bearing (S)-MeO-Biphep ligand. This methodology is thus of high interest for its potential application in the preparation of optically pure terutroban in particular and 2-aminotetralines in general.
Acknowledgments
This work was supported by Oril Industries (A.M.M.). The authors thank J.-P. Lecouvé, L. Vaysse-Ludot for fruitful discussions.