

1 Introduction
Catalytic hydrogenation is a process widely used in petrochemistry and transformation of specialty chemicals [1,2]. In many applications, selective hydrogenation is required. This contribution presents a review of some highlights published during this last decade for selective hydrogenations in liquid phase over bimetallic catalysts synthesized in our research team. The works detailed in this paper allow appreciating the influence of the bimetallic systems formulation on the catalytic performances, and notably put emphasis on the use of redox processes for catalyst preparation. Two applications of selective hydrogenation are detailed: citral hydrogenation and nitrate reduction.
Numerous natural compounds issued from biomass contain both CC and CO bonds, like in α,β-ketoalkenes, more specifically in unsaturated aldehydes as citral (3,7-dimethyl-2,6-octadienal). In many applications, selective hydrogenation is required. Monooxygenated terpenes are an important class of natural compounds whose transformation may require selective CO hydrogenation while preserving the CC double bonds [3,4]. Metal catalysts containing noble metals (e.g., Pt, Pd) are known to be active in CC bond hydrogenation and thus poorly selective to unsaturated alcohols from unsaturated aldehydes. To increase selectivity, several strategies such as addition of a second metal [5], use of reducible supports [6], use of selective poisons [7] or manipulation of solvent effect [8], etc., can be employed.
Catalytic hydrogenation is also a possible process to treat polluted waters by reducing nitrate ions into nitrogen. The reaction proceeds in two steps: (i) reduction of nitrate to nitrite ions, and (ii) reduction of nitrites to N2 [9]. Bimetallic catalysts are necessary for the reaction to occur at a reasonable rate at room temperature: while certain noble metals (Pd, Pt) are very active in nitrite hydrogenation, they cannot perform alone the initial reduction step of nitrate to nitrite. A second metal should be added, allowing the complete reduction of nitrate to N2.
Various parameters involved in the formulation of the bimetallic catalysts will be studied and their influence on the catalytic performances for the two studied selective hydrogenation reactions will be presented. In spite of very different conditions (moderate temperature, high pressure, organic solvent for citral hydrogenation; room temperature, atmospheric pressure and reaction in water for nitrate hydrogenation), some rules will be proposed on the basis of a critical comparison between the two processes.
2 Experimental
2.1 Catalyst preparation
For the catalysts used in citral hydrogenation, γ-Al2O3 (surface area 240 m2 g−1), TiO2 (Degussa P25, surface area 50 m2 g−1) and SiO2 (Degussa, surface area 180 m2 g−1) were used as supports with particles sizes between 0.10 and 0.04 mm. Supported 1 wt% Rh/Al2O3, 1 wt% Rh/TiO2 and 1 wt% Pd/SiO2 monometallic catalysts were prepared by ion exchange in aqueous medium using RhCl3 as the precursor salt and by wet impregnation using a solution of Pd(acac)2 in acetone, respectively. The samples were further calcined at 300 °C and reduced under hydrogen flow at 300 °C or 500 °C, generally. Rh–Ge/Al2O3 and Rh–Ge/TiO2 bimetallic catalysts (with various Ge contents) were prepared by surface redox reaction between hydrogen activated on the Rh particles surface of the parent catalyst, and the Ge salt (GeCl4) dissolved in water (“catalytic reduction” method, named CR), according to a procedure described elsewhere [5]. Otherwise, some Rh–Ge/Al2O3 and Pd–Sn/SiO2 bimetallic catalysts were prepared by successive impregnations (SI) via the impregnation of the Ge or Sn precursor on the parent monometallic catalyst. After drying, all the bimetallic samples (CR and SI) were reduced under hydrogen at the same temperature as that used for the parent catalyst. The elemental analysis of the metallic function contents (wt%Rh, wt%Ge, wt%Sn) was performed by atomic absorption.
For catalysts used for nitrate reduction, monometallic catalysts were prepared by an impregnation method using aqueous solutions of the precursor salt, Pd(NH3)4(NO3)2 or PdCl2. After evaporation of water, the catalysts were dried at 120 °C. For metal oxide supports (silica, alumina, and ceria), the catalysts were then calcined in flowing air (80 mL min−1) at 400 °C (6 °C min−1) for 4 h, and cooled to room temperature under N2. Finally the catalysts were reduced by flowing hydrogen (60 mL min−1) at 400 °C (6 °C min−1) for 6 h. For the other supports (activated carbon and organic polymers), the catalysts were directly reduced, but only at 100 °C for those deposited on organic polymers. Bimetallic catalysts were prepared by a controlled surface reaction, favouring metal–metal interactions, or successive impregnation.
2.2 Temperature programmed reduction (TPR)
In some cases, TPR experiments were performed in order to investigate the hydrogen consumption during the final reduction step of the catalysts. Thus, the TPR were conducted on catalysts as prepared, dried at 100 °C under hydrogen and transferred to the TPR reactor under inert atmosphere in order to avoid the oxidation by air, in which the temperature was raised from 25 °C to 500 °C with a ramp of 5 °C min−1 under a 1.0 vol.% H2/Argon gas mixture. The hydrogen uptake was monitored by a thermal conductivity detector.
Otherwise, TPR experiments were performed on samples pretreated in situ under oxygen for 30 min at 300 °C and cooled down to room temperature. The conditions of the reduction under programmed temperature remained the same as above.
2.3 Fourier Transform Infrared (FTIR) spectroscopy of adsorbed CO
Transmission FTIR spectra were collected in the single beam mode, with a resolution of 2 cm−1, using a Nicolet Nexus 470 FT-IR spectrometer equipped with an MCT-B detector, according to a procedure well described elsewhere [10]. Prior to CO-adsorption experiments, all catalysts were reduced in situ under H2 at their initial reduction temperature, then flushed with inert gas at this temperature before to be cooled to room temperature. At room temperature, a flowing CO/He mixture was introduced in the cell and spectra were collected until a steady state was reached. Spectral deconvolution was performed using the Galactic PeakSolve peak-fitting program (correlation factors above 0.999 and standard errors below 0.004).
2.4 X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy (XPS) measurements were collected using a hemispherical analyzer on a Kratos Axis Ultra DLD XPS with a monochromated Al Kα X-ray source. In a pretreatment cell directly attached to the XPS chamber, the catalysts were pretreated in situ in H2 atmosphere during 1 h at the same temperature as during their initial reduction. The samples were then cooled to room temperature and moved without exposure to the air into the ultra-high-vacuum chamber (10−9 torr). Peak fitting was achieved using XPSPeak 4.1 software. For the analysis of Pd–Sn/SiO2 catalysts, binding energies were calibrated with respect to Si 2p (103.3 eV) and were then modelled using a mixed Shirley–Linear background. Elemental surface stoichiometries were obtained from peak area ratios corrected by appropriate sensitivity, by using Gaussian/Lorenzian sum functions to fit the line shapes.
2.5 Catalytic tests
2.5.1 Citral hydrogenation
Liquid-phase hydrogenation of citral was carried out in a 300-mL stirred autoclave (Autoclave Engineers, fitted with a system for liquid sampling) either at 70 °C (Rh-based catalyst) or 130 °C (Pd-based catalysts), and at constant pressure of 7 MPa. Prereduced catalysts were immersed into 90 mL of solvent (isopropanol 99%) without exposure to the air before introduction into the autoclave. After a first flush with nitrogen and a second one with hydrogen, the temperature was raised to 70 °C or 130 °C under 3 MPa of hydrogen. Then a mixture of substrate (3 mL of citral) and of isopropanol (10 mL) was loaded into the autoclave through a cylinder under a 7-MPa hydrogen pressure. Zero time was taken at this moment and stirring was switched on. Liquid samples were analysed by gas chromatography on a Varian 3400 chromatograph provided with a FID detector and a capillary column DB-WAX (J&W, 30 m, 0.53 mm i.d.) using nitrogen as a carrier gas.
2.5.2 Nitrate reduction
The nitrate reduction reaction was performed in a semi-batch reactor, at atmospheric pressure and 25 °C or 30 °C. In a typical experiment, 64 mg of the powdered catalyst was used. The catalyst was first reduced under hydrogen (100 °C; 0.5 h). During this time, 90 mL of ultra-pure water (18.2 MΩ) were placed in a vial purged with nitrogen. When the catalyst was stabilized in temperature, the ultra-pure water was added over the reduced catalyst. Then, the catalyst was maintained in water under bubbling of hydrogen or of a hydrogen/carbon dioxide mixture. Afterwards, 10 mL of a solution (16 mmol L−1) of nitrate (KNO3) were introduced in the reactor to start the reaction. The catalyst's dispersion in the aqueous medium was achieved by gas flowing (flow rate = 250 mL min−1) through a porous glass located at the bottom of the reactor. To monitor the progress of the reaction, representative aqueous samples were periodically taken out and immediately separated by filtration and then analyzed by HPLC.
3 Results and discussion
Various bimetallic catalysts were evaluated for the two studied hydrogenation reactions in liquid phase, namely nitrate reduction in water and citral hydrogenation in isopropanol. Nitrate reduction principally yields dinitrogen, with ammonia as a by-product and nitrite as an intermediary product. The reduction of citral is more complex and can lead to a variety of products. Fig. 1 presents the main reaction pathways that can occur during this reaction. A first step is the reduction of either the CO or the conjugated CC bond to produce geraniol and nerol (UA: unsaturated alcohols) or citronellal, respectively. Consecutive hydrogenation leads to citronellol and finally to 3,7-dimethyloctanol. Apart from these reactions, secondary processes of cyclization or of reaction with the solvent (alcohol) can lead to other by-products like isopulegol or acetals of citronellal.
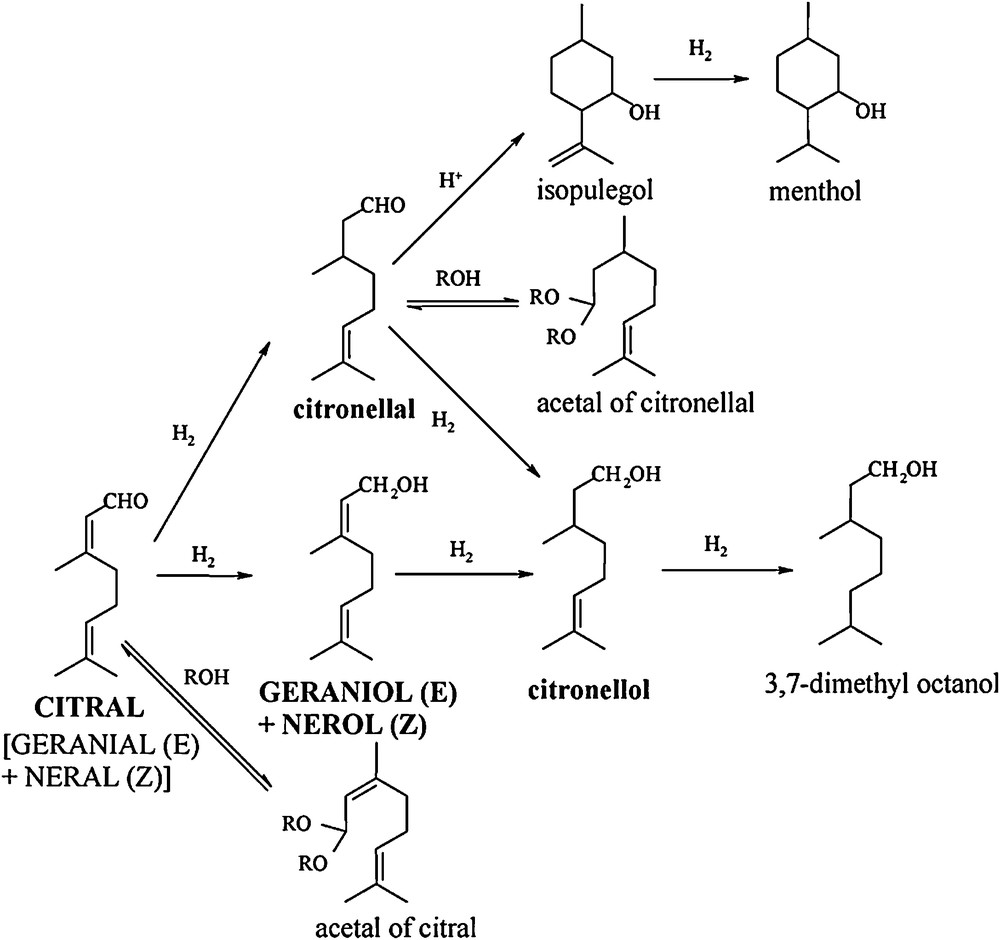
Reaction scheme for citral hydrogenation.
3.1 Preparation of bimetallic catalysts by surface redox reaction
In the course of the preparation of supported bimetallic catalysts, the main difficulty is to create an intimate contact between the two metals in order to obtain bimetallic entities. Classical methods such as co-impregnation (CI) or successive impregnations (SI) often prove to be unsatisfactory, since they are not always reproducible and do not lead necessarily to the formation of bimetallic species. Among the techniques allowing the optimisation of the interaction between the two metals, surface redox reactions proceed in two steps: firstly, a parent monometallic catalyst is prepared and further modified in liquid phase by a redox reaction between its metallic surface particles (or a reducer adsorbed on its surface) and the oxidized form of the modifier introduced in solution [11–14]. This preparation method favours the formation of bimetallic particles, compared to classical techniques such as successive impregnation, for which a random distribution of the second metal on the parent monometallic catalyst is generally observed. For example, when prepared by redox reaction, namely catalytic reduction (CR), bimetallic Pt–Cu/Al2O3 catalysts present roughly 80% of bimetallic particles with the same Cu/Pt molar ratio as the nominal one, whereas on the same catalyst prepared by successive impregnation, metal particles present various platinum and copper contents, between 0 and 100 mol% [15].
The preparation method plays a direct role in the bimetallic particle composition, and consequently has a strong impact on the catalytic performances. Thus, a series of Rh–Ge/Al2O3 bimetallic catalysts were prepared by catalytic reduction (CR), reduced at 300 °C and tested for the citral hydrogenation in isopropanol as a solvent. As reported elsewhere [5,16], the presence of bimetallic particles on these systems was revealed by transmission electron microscopy (TEM) analysis coupled with energy dispersive X-ray (EDX) spectroscopy, as well as by TPR experiments performed to investigate the hydrogen consumption during the final reduction of the catalysts. Fig. 2a displaying citral conversion as a function of time obtained with the Rh–Ge/Al2O3 bimetallic catalysts shows a rapid hydrogenation of citral during the first few minutes, followed by an activity decrease after this earlier period (this decrease being in a lower extent for the highest Ge content). This phenomenon is generally explained by a decomposition of the citral or unsaturated alcohols yielding chemisorbed CO and carbonaceous species that accumulate on the catalysts’ surface and block a part of the active sites [17–24]. According to Fig. 2b, the selectivity of unsaturated alcohols (UA) increases with the Ge content in the bimetallic samples, going from 2% for the 1.0 wt% Rh/Al2O3 monometallic catalyst (not reported) until 75% for the 1.0 wt% Rh–3.5 wt% Ge/Al2O3 sample. Otherwise, no significant evolution of UA selectivity is observed as a function of citral conversion. FTIR spectra of adsorbed CO on these catalysts are shown in Fig. 3, and the corresponding peak assignments and the relative areas under each peak obtained after decomposition are summarized in Table 1. Similar spectral features are observed for the bimetallic and monometallic Rh samples, no CO adsorption occurring on a Ge/Al2O3 monometallic catalyst. However, the different surface CO species on the bimetallic catalysts are affected by the presence of Ge: (i) the bridged species, as well as species assigned to Rh0x, decrease (the peak corresponding to bridge-bonded CO species on Rh0 even totally disappears for the highest Ge contents), (ii) the gem-symmetric species on Rh1+ becomes dominant on the bimetallic catalysts, (iii) a new peak at high frequency (approximately 2130 cm−1) appears on the spectra of the bimetallic catalysts containing the two highest Ge contents (noticed as a shoulder in the original spectra), and is assigned to CO linearly bonded on Rh2+ and/or Rh3+ ions [25–27]. All these observations were explained by the fact that Ge gradually covers the Rh surface, creating smaller Rh1+ ensembles at the expense of larger Rh0 surfaces, and that a charge transfer is generated from Rh to Ge, more noticeable at higher Ge loadings [10].
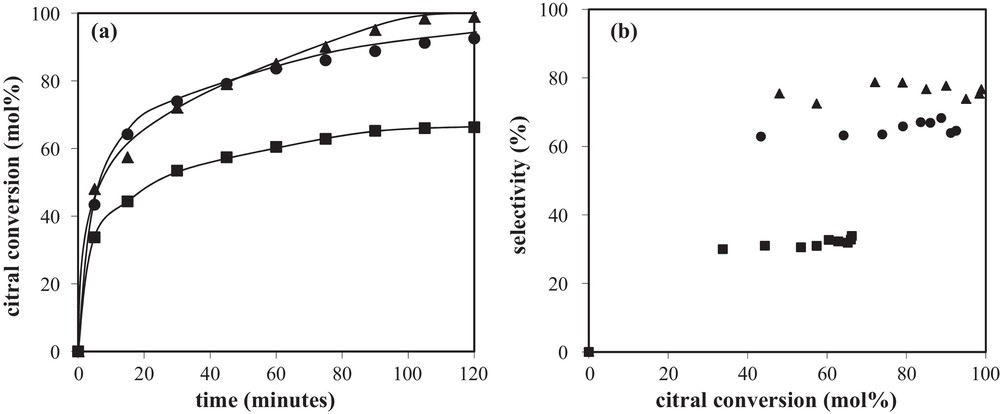
(a) Citral conversion as a function of time and (b) selectivity to unsaturated alcohols as a function of citral conversion obtained with Rh–Ge/Al2O3 bimetallic catalysts prepared by catalytic reduction and reduced at 300 °C: () 1 wt%Rh–1 wt%Ge/Al2O3; () 1 wt%Rh–2.5 wt%Ge/Al2O3; () 1 wt%Rh–3.5 wt%Ge/Al2O3.
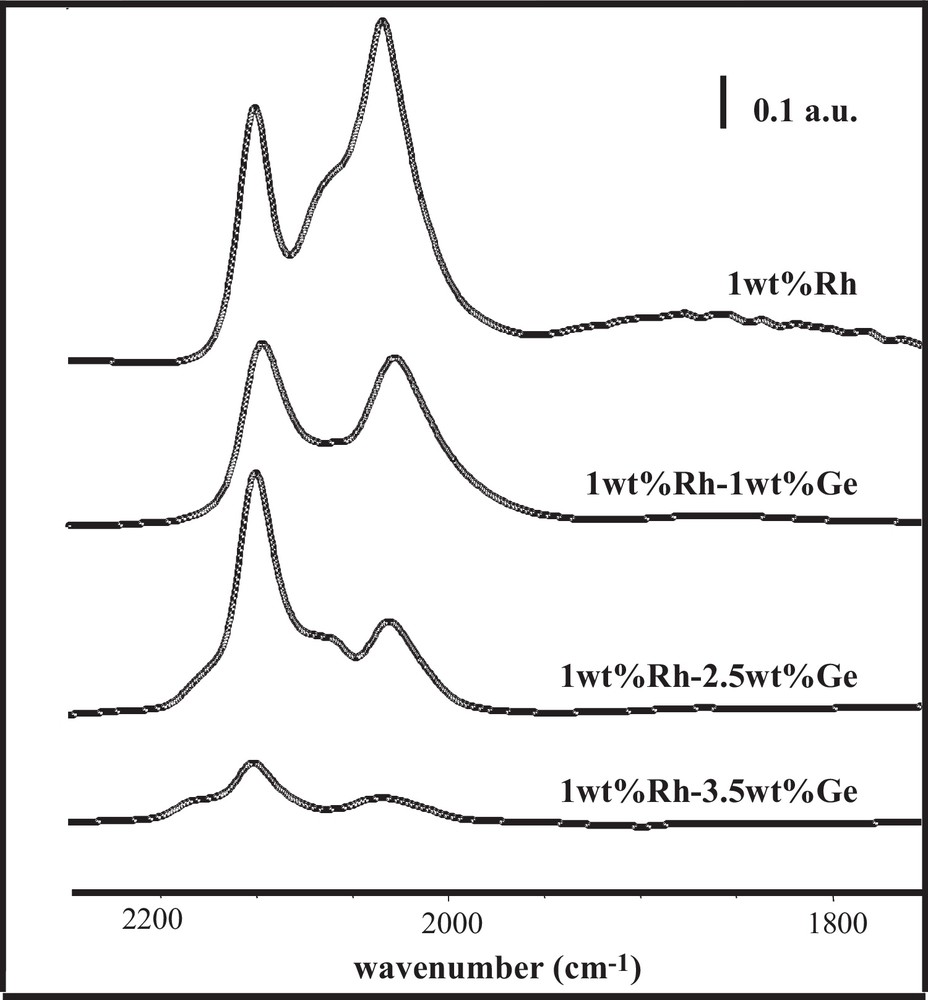
FTIR spectra of adsorbed CO on the Rh/Al2O3 monometallic catalyst and the Rh–Ge/Al2O3 bimetallic catalysts prepared by catalytic reduction and reduced at 300 °C.
Peak assignments (wavenumber, cm–1) and relative surface areas (%) under each peak observed during CO adsorption followed by FTIR on bimetallic Rh–Ge/Al2O3 catalysts prepared by catalytic reduction and reduced at 300 °C.
| Catalyst sample | Bridged on Rhx0 | Bridged on Rh1+ | Gem asym on Rh1+ Rh(CO)2 | Linear on Rhx0 RhCO | Gem sym on Rh1+ Rh(CO)2 | Linear on Rh>1+ ORhCO |
| 1 wt%Rh | 1865 4.6% | 2025 20.6% | 2034 30.1% | 2065 21.8% | 2101 22.9% | — |
| 1 wt%Rh–1 wt%Ge | 1854 2.5% | 2017 23.0% | 2028 20.2% | 2074 19.2% | 2098 35.1% | — |
| 1 wt%Rh–2.5 wt%Ge | — | 2018 13.4% | 2034 15.5% | 2067 18.3% | 2101 50.6% | 2129 2.2% |
| 1 wt%Rh–3.5 wt%Ge | — | 2016 8.1% | 2038 12.9% | 2085 11.3% | 2102 51.6% | 2132 16.1% |
Finally, during citral hydrogenation, the effect of the Ge modifier deposited in interaction on the Rh particles was attributed, firstly, to an inhibition of the ethylenic function adsorption in order to avoid citronellal formation, while keeping accessible surface metallic sites for hydrogen activation, and secondly, to an activation of the citral carbonyl group by interaction between the lone electron pair of its oxygen and Gen+ species (acting as Lewis acid sites) [28–30]. With this example, the interest of using surface redox reactions to control the deposit of the additive on the parent catalyst surface is then highlighted.
3.2 Comparison of bimetallic catalysts prepared by various methods
Monometallic catalysts supported on alumina or silica are not active for nitrate reduction. For this reaction, it is necessary to associate (i) a noble metal, such as palladium or platinum with (ii) a transition metal such as copper, tin or indium, for example. It was shown that the interaction between the noble metal and the promoter is a key parameter for this reaction. Bimetallic Pt–Cu/Al2O3 catalysts with the same nominal composition were prepared by adding copper onto a monometallic Pt/Al2O3 catalyst by (i) a redox reaction (catalytic reduction, CR) favouring the deposition of copper onto platinum and (ii) successive impregnation (SI) by cationic exchange with the support favouring the deposition of copper onto alumina [15]. The most active catalyst was the one prepared by catalytic reduction, which was able to completely convert nitrates in 30 min, whereas the total conversion was not reached with the one prepared by successive impregnation [15]. In bimetallic catalysts, the role of the promoter, i.e. copper in the present systems, is to reduce nitrate into nitrite according to a redox process. During this step, the interaction between the promoter and the noble metal is of major importance to maintain the promoter in the metallic state via hydrogen adsorbed on the noble metal. This mechanism involves that the promoter should be easily oxidized by nitrate in the reaction conditions and then reduced by hydrogen chemisorbed on the noble metal.
The preparation method also strongly influences the catalytic properties of bimetallic catalysts for citral hydrogenation. Fig. 4 displays the comparison of the catalytic properties for this reaction of two Rh–Ge/Al2O3 catalysts reduced at 300 °C, but prepared either by catalytic reduction method (CR) or by conventional impregnation of Ge salt on a Rh parent catalyst (i.e. successive impregnation method, SI). For a same Ge content (i.e. 2.5 wt%Ge in the presented example), the CR catalyst is much more active than the SI one. The UA selectivity (very slightly dependant of the citral conversion) is quite comparable between the two samples, but the proportion of geraniol and nerol in the reaction mixture after a 2-h reaction time is smaller on the SI catalyst compared to the CR one (30 mol% instead of 60 mol%, because of the lower activity with the SI sample). Thus, the preparation method of supported Rh–Ge bimetallic catalysts plays a prominent role on their catalytic properties for citral hydrogenation. The higher performances of the CR catalyst in comparison with the SI sample can be explained by a deeper modification of the Rh properties by Ge in the former, highlighted otherwise by a more noticeable decrease of the Rh activity after Ge introduction for structure insensible catalytic reactions such as cyclohexane dehydrogenation and toluene hydrogenation [5].
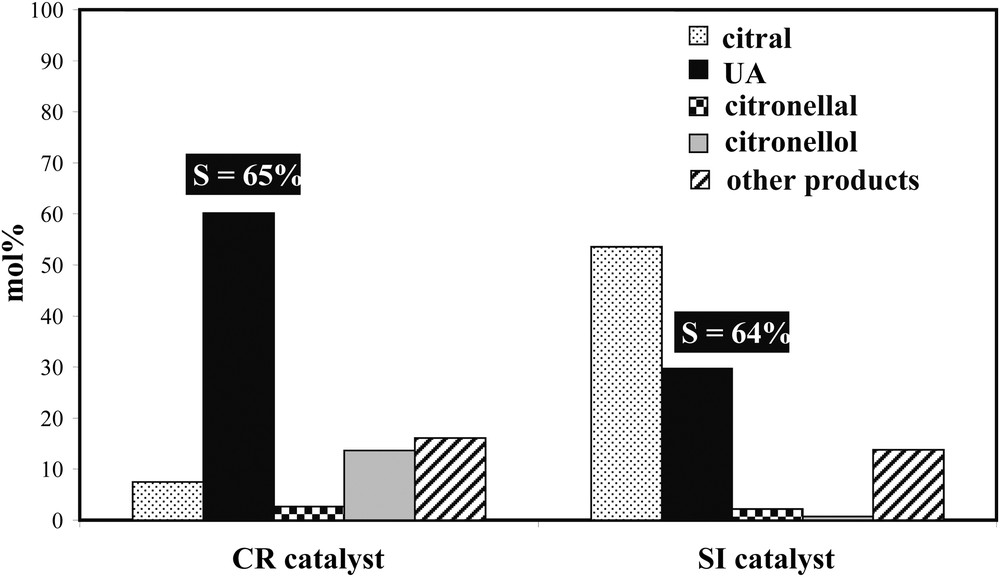
Citral hydrogenation on 1 wt%Rh–2.5%Ge/Al2O3 catalyst prepared by catalytic reduction (CR) or successive impregnations (SI), reduced at 300 °C: composition of the reactional mixture and UA selectivity after a 2-h reaction time.
These examples highlight that the creation of an intimate contact between the additive and the metallic particles of the parent catalyst by using surface redox reactions significantly contributes to improve the catalytic performances. The interest of the surface redox reactions for the preparation of bimetallic catalysts was also observed for other catalytic systems and applications [12,14,31,32].
3.3 Influence of the activation treatment of CR catalysts
The catalytic properties are strongly influenced by the activation of the catalysts, which also may lead to a sintering or a segregation of the metals.
3.3.1 Influence of the nature of the treatment
The influence of different gas treatments after addition of the second metal by redox reactions was examined. Pt–Cu/Al2O3 catalysts prepared by catalytic reduction were tested for nitrate reduction either directly after drying overnight at 70 °C under nitrogen flow or after air exposure at ambient temperature further the drying step [33]. The nitrate evolution vs. time obtained for both experiments is presented in Fig. 5. The most active catalyst is the one that has been in contact with air, which involves a migration of copper initially deposited onto platinum particles, thus increasing Pt accessibility in bimetallic catalysts. Without this exposure to the air, the coverage of Pt particles by Cu is probably too important. Conversely, a poor activity was obtained when the Pt–Cu/Al2O3 sample was oxidized by calcination at 400 °C, the segregation between the two metals being too important with a migration of copper towards the alumina support. The same catalyst reduced at 400 °C under pure hydrogen for 1 h after contact with the air showed a lower activity than the one observed without any pretreatment, which was explained by a diffusion of copper inside the platinum particles and an enrichment of platinum in surface during the reduction step [33].
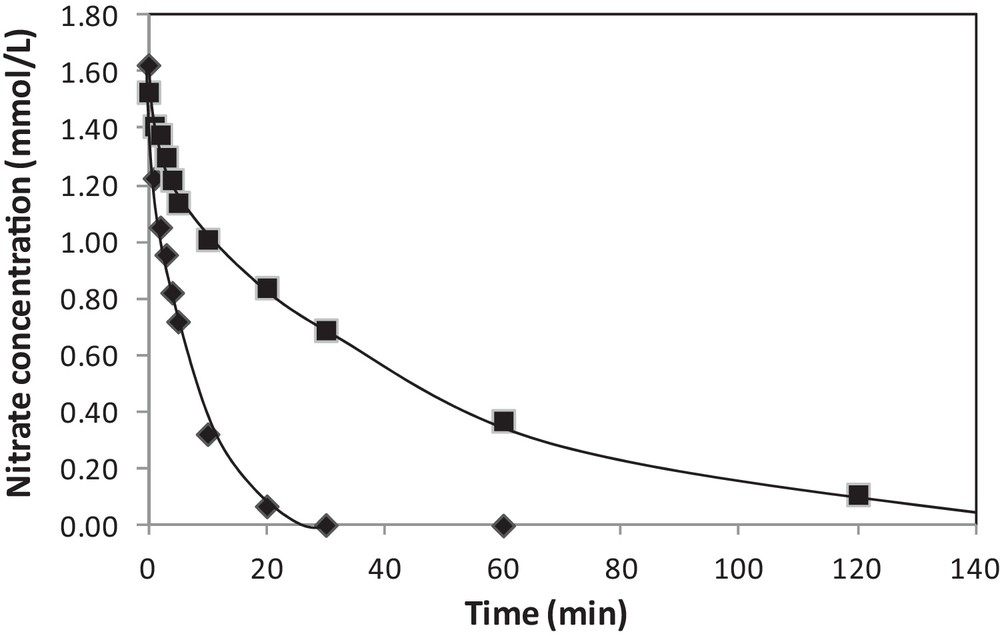
Effect of the contact with the air on the activity of 3 wt%Pt–1 wt%Cu/Al2O3 prepared by redox reaction, with () or without () contact with the air after preparation (initial nitrate concentration: 1.6 mmol L−1, T = 10 °C, PH2 = 1 bar).
3.3.2 Influence of the reduction temperature
More often, after preparation by catalytic reduction and drying, catalysts are directly reduced under hydrogen at various temperatures. Fig. 6, displaying the citral conversion and the unsaturated alcohols selectivity as a function of the reduction temperature of Rh–Ge/Al2O3 bimetallic catalysts prepared by catalytic reduction (CR), shows that the hydrogenating properties are affected by the activation treatment conditions under hydrogen. The shape of the citral conversion curves (Fig. 6a) depends on the Ge content: for the 1 wt% Ge content, the 60 min citral conversion increases in a monotonous way with the temperature until a plateau between 400 °C and 500 °C; for higher Ge contents (2.5 wt% and 3.5 wt% Ge), the conversion increases between 150 °C and 300 °C, then drastically decreases at higher temperatures to reach a low value at 500 °C. In order to follow hydrogen consumption in the course of the final reduction of bimetallic catalysts, the different samples, as well as Rh- and Ge-supported monometallic samples, were characterized by temperature programmed reduction, starting from samples dried overnight at 100 °C under hydrogen flow after their preparation by the CR method (Fig. 7). The Ge addition strongly modifies the shape of the TPR curve of the Rh parent catalyst. The first peak near 30 °C decreases as the Ge content increases, in accordance with Ge deposition on Rh particles, since this peak is assigned to hydrogen adsorption on Rh0 surface (as shown in Fig. 7, Ge is not able to activate hydrogen at ambient temperature). On bimetallic catalysts, this peak is followed by H2 consumption in the 40 °C–120 °C temperature range, which decreases with the increase in the Ge content, and is attributed to hydrogen adsorption on Rh–Ge species containing few Ge atoms. In the 120–400 °C temperature range, the hydrogen consumption of bimetallic catalysts is higher than that of the monometallic Rh one and increases with the Ge content, meaning that oxidized Ge atoms are reduced. Otherwise, the reduction of these Ge species is promoted by Rh, since the reduction of the monometallic Ge catalyst starts only at 400 °C. Finally, hydrogen consumption in the 400–500 °C temperature range corresponds to the reduction of oxidized Ge species isolated on the alumina support, as for the monometallic Ge catalyst. Then, in the course of the preparation of Rh–Ge/Al2O3 catalysts by catalytic reduction, Ge deposition occurs on the Rh particles and also partly on the support [5].
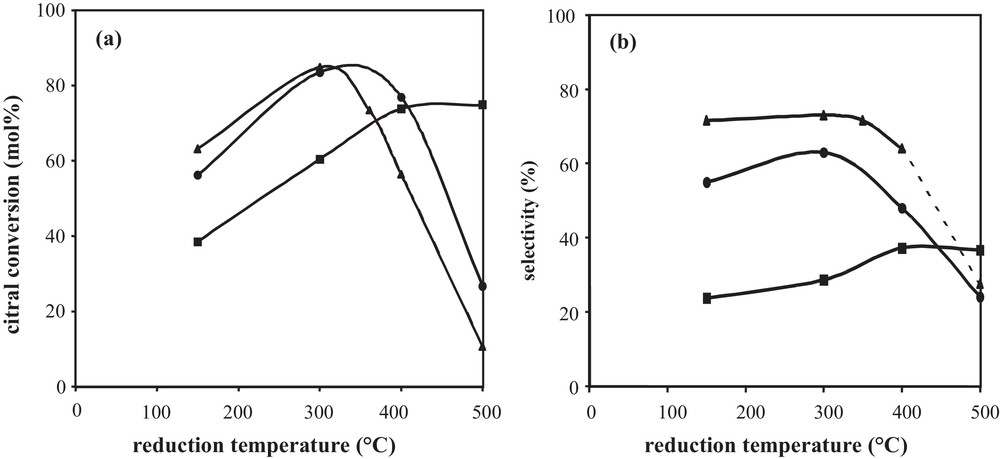
(a) Citral conversion after 60 min reaction time and (b) selectivity to unsaturated alcohols at 40% citral conversion according to the reduction temperature of Rh–Ge/Al2O3 bimetallic catalysts prepared by catalytic reduction: () 1 wt%Rh–1 wt%Ge/Al2O3; () 1 wt%Rh–2.5 wt%Ge/Al2O3; () 1 wt%Rh–3.5 wt%Ge/Al2O3.
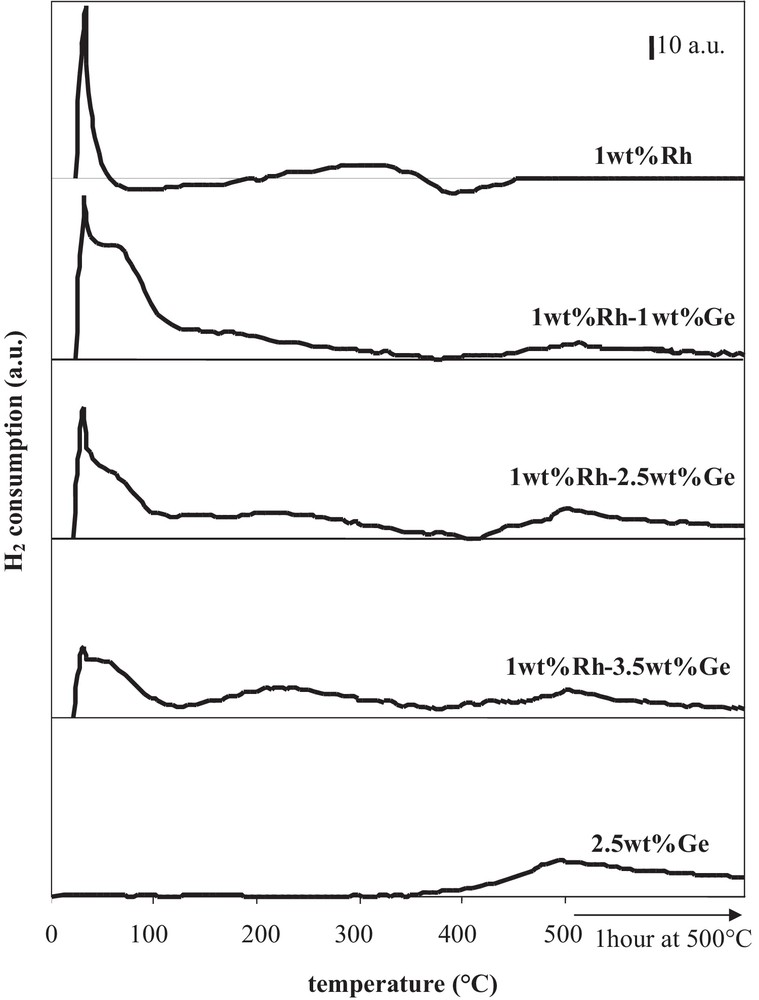
TPR profiles of Rh/Al2O3 and Ge/Al2O3 monometallic catalysts, and of Rh–Ge/Al2O3 bimetallic catalysts prepared by catalytic reduction.
From the TPR profiles shown in Fig. 7, an increase in the reduction temperature of the bimetallic catalysts from 150 °C to 300 °C leads to the reduction of Ge species promoted by Rh, justifying the catalyst activity enhancement obtained in this range of temperature for all samples reported in Fig. 6a. Furthermore, in the 400–500 °C temperature range, the reduction of oxidized Ge species deposited on the support induces different effects depending on the Ge content. In the case of the 1 wt% Rh–1 wt% Ge/Al2O3 catalyst, the reduction of these Ge species leads to a more active catalyst for citral hydrogenation, due to a Ge enrichment of bimetallic particles and in agreement with the enhancement of the conversion when the Ge content goes from 1 wt% to 2.5 wt% (Fig. 2a). But at higher Ge contents (2.5 and 3.5 wt% Ge), the reducing treatment until 500 °C leads to the reduction of more Ge species deposited on the support, which induces an inhibiting effect on catalytic activity (Fig. 6a).
The selectivity to unsaturated alcohols follows the same evolution as the conversion when increasing the temperature from 150 to 500 °C (Fig. 6b). However, the increase in the UA selectivity is less significant than that of the conversion, which means that the activity enhancement is not only related to the presence of new active sites for the hydrogenation of the carbonyl function. In addition, the drastic drop of the selectivity to unsaturated alcohols after a reduction at 500 °C arises mainly from the formation of secondary products and not from a preferential hydrogenation of the conjugated CC bond leading to citronellal.
Consequently, these results show that the activation treatment of the bimetallic catalysts prepared by surface redox reactions is crucial, and modulates the interaction degree and respective localization of the modifier and parent metal species on the catalyst surface.
3.4 Influence of the nature of the support
For nitrate reduction, monometallic palladium catalysts modified by addition of copper, tin or indium are very active. Nevertheless, it was observed that the support plays also an important role during this reaction. Indeed, hydroxide ions are produced during the reduction process, leading to an increase of pH, which has been demonstrated as detrimental to obtain a low selectivity to ammonia, the undesired product. Thus, the support may play a role in this reaction since it may help to control the pH near the active centres and it also influences the metal dispersion and metal–metal interaction. Nevertheless, depending on the support, an external control of the pH is often necessary and can be done by adding carbon dioxide to the hydrogen flow. Table 2 compares the best results obtained in roughly similar conditions (T = 25–30 °C, initial nitrate concentration = 1.6 mmol L−1, mass of catalyst = 640 mg L−1, gaseous flow rate = 250 mL min−1, atmospheric pressure) with Pd–Sn and Pd–Cu catalysts with optimized metal contents on various supports. For metal oxides’ support (CeO2, Al2O3 and SiO2), the addition of CO2 in the gas phase is necessary to control the pH of the solution at values lower than 5–6 and to reach selectivities to ammonium ions lower than 10–15%. In the absence of CO2, 45% of nitrates are converted to ammonium ions on the alumina supported Pd–Sn catalyst. For this reason, other types of supports with capabilities of neutralizing the hydroxide ions produced in the course of nitrate reduction were used: (i) a carbon support obtained from grape seeds and activated with phosphoric acid, (ii) a sulfonated poly(styrene-divinylbenzene) support (also acidic), and (iii) two conducting polymers, polyaniline and polypyrrole, with anion exchange properties. The use of these non-usual supports allowed performing nitrate reduction in the absence of external pH control and the performances of the resulting Pd–Sn catalysts were higher than the ones obtained with classical metal oxides supports.
Activity for nitrate reduction and selectivity to ammonia of Pd–Cu and Pd–Sn catalysts with optimized composition and metal contents on various supports (T = 25–30 °C, initial nitrate concentration = 1.6 mmol L–1, mass of catalyst = 640 mg L–1, gaseous flow rate = 250 mL min–1, atmospheric pressure).
| Catalyst compositiona | Reaction temperature (°C) | Gas phase composition | Ab (μmol min–1gcata–1) | Sc (%) | Ref. |
| 5Pd–1.5Sn/Al2O3 | 25 | H2 + CO2 (50/50) | 49 | 7.2 | [34] |
| 5Pd–2Sn/Al2O3 | 25 | H2 | 32 | 45 | [35] |
| 5Pd–1.5Sn/SiO2 | 25 | H2 + CO2 (50/50) | 34 | 6.8 | [34] |
| 5Pd–1.75Sn/CeO2 | 25 | H2 + CO2 (80/20) | 8.2 | 9.5 | [36] |
| 5Pd–2.5Cu/GSd | 30 | H2 | 5.2 | 1.9 | [37] |
| 5Pd–2Sn/Poly(Sty–DVB)e | 25 | H2 | 5 | 13 | [38] |
| 5Pd–0.5Sn/PPyf | 25 | H2 | 150 | 4.8 | [35] |
| 5Pd–1Sn/PANIg | 25 | H2 | 47 | 1.8 | [35] |
a Metal content in wt%.
b Global activity for nitrate reduction.
c Final selectivity to ammonium ions.
d Activated carbon from grape seeds.
e Poly(styrene-divinylbenzene).
f Polypyrrole.
g Polyaniline.
For citral hydrogenation, the support may also play a role in the performances of the bimetallic catalysts. Table 3 compares the catalytic properties for citral hydrogenation of 1 wt%Rh–1 wt%Ge bimetallic catalysts prepared by catalytic reduction and supported either on Al2O3 (catalysts previously described in Sections 3.1 and 3.3.2) or TiO2 (reducible oxide). As previously observed on the alumina support, the presence of bimetallic particles was also put in evidence on the titania support by several characterization techniques (TEM, TPR, probe reaction of cyclohexane dehydrogenation catalysed by the Rh metallic function) [39]. For the same reduction temperature (300 °C), TiO2 support leads to a bimetallic catalyst more active and more selective towards unsaturated alcohols, highlighting the influence of the nature of the support on the characteristics of the generated bimetallic particles. Particularly, the average metallic particle size is lower on TiO2 compared to Al2O3, and the Ge enrichment in the bimetallic particles may also differ according to the nature of the support. Otherwise, the different behaviour of the two Rh–Ge bimetallic catalysts according to the support nature is even more noticeable after reduction at 500 °C: with TiO2, the increase in the reduction temperature from 300 to 500 °C leads to an decrease of the citral conversion and a marked increase of the UA selectivity. The results obtained on TiO2 support compared to Al2O3 were attributed to its reducible properties, and the creation of TiO(2–x) partially reduced species after reduction at 500 °C, which block some Rh active sites and modify the CC/CO adsorption competition in the course of citral hydrogenation [40].
Citral conversion after a 15-min reaction time and selectivity to unsaturated alcohols (UA) at 60% citral conversion on bimetallic 1 wt%Rh–1 wt%Ge catalysts prepared by catalytic reduction and supported on various oxides (Al2O3 or TiO2).
| Catalyst | Treduction (°C) | Citral conversion (mol%) | UA selectivity (%) |
| Rh–Ge/Al2O3 | 300 500 | 43 60 | 30 39 |
| Rh–Ge/TiO2 | 300 500 | 54 30 | 55 78 |
3.5 Presence of alloy
The study concerning the effect of Sn addition to Pd catalysts on the citral selective hydrogenation to unsaturated alcohols constitutes a representative example of the alloy's role [41]. Indeed, Sn addition to Pd/SiO2 catalysts by successive impregnation method (reduction temperature = 300 °C) significantly modifies their catalytic properties, inducing a great promoting effect on UA selectivity (Table 4). As it is well known from the literature, Pd/SiO2 monometallic catalysts are very active for citral hydrogenation, leading exclusively to the formation of CC hydrogenated products and then to an UA selectivity equal to 0 [18,42,43]. For the bimetallic catalysts, the addition of a low Sn content (≤ 1 wt%) slightly modifies the conversion of the monometallic catalyst, but for the highest Sn contents (> 1 wt%), a drastic conversion drop is observed. On the contrary, the UA selectivity strongly increases with the Sn content, reaching a maximum for 1.5 wt% Sn content. This promoting effect was related to the structure of the bimetallic particles, which were fully characterized by TEM analysis coupled with EDX, TPR, FTIR of adsorbed CO and XPS [41]. The curve-fitted XPS spectra of the Pd 3d and Sn 3d regions for the monometallic and bimetallic catalysts are presented in Fig. 8, leading to the data gathered in Table 4 for the two 3d5/2 core levels. XPS analysis is known to give atomic compositions representative of the material surface until 10 nm in depth, which was considered to be representative of the bulk composition in the case of these samples presenting average particle sizes between 5.7 and 8.0 nm. In Fig. 8A, only one band is obtained in the 3d5/2 area of the Pd core level, since all Pd atoms are in the Pd0 reduced form after reduction (as clearly demonstrated otherwise by TPR experiments) [41]. The full-width-half-maximum (FWHM) of the Pd 3d5/2 peak given in Table 4 decreases with increasing the Sn content, which implies an increase of the Sn quantity in contact with Pd [44]. Nevertheless, the FWHM obtained for bimetallic catalysts with Sn content ≥ 1.5 wt% are very close, suggesting that the Sn quantity in contact with Pd reaches a plateau. In agreement with the literature [24,44–47], the Sn 3d5/2 energy peak is decomposed into two contributions: a first one at 485.0 eV corresponding to oxidized Sn (Sn2+ and/or Sn4+, without possible discrimination between the two forms), and the second one at lower binding energy (482.0 eV) assigned to reduced Sn. On all reduced bimetallic catalysts, oxidized Sn species are the prevailing ones (Fig. 8B), with however an increase of the Sn0/Sntotal atomic ratio with the Sn content (Table 4). From TPR results, the reduction temperature (300 °C) was shown as not enough elevated to reduce Sn species isolated on the support [41]. Consequently, the quantity of reduced Sn0 species deduced from XPS is inevitably in contact with Pd, leading to an estimation of 0.37 wt% Sn0 and 0.34 wt% Sn0 for the 1 wt% Pd–1.5 wt% Sn/SiO2 and 1 wt% Pd–2 wt% Sn/SiO2 catalysts, respectively. Moreover, the binding energy of the Sn0 3d5/2 peak increases with the Sn content (Table 4), indicating an electron deficiency on Sn due to an electron transfer from Sn to Pd [45,47], as highlighted also by FTIR results obtained on these samples [41]. Finally, the formation of an alloy with a Pd3Sn composition accompanied by an electron transfer from Sn to Pd was suggested for the Pd–Sn/SiO2 bimetallic catalysts with Sn content superior to 1.5 wt%. Such alloyed phase was considered as responsible for the maximum UA selectivities (> 75%).
Catalytic performances during citral hydrogenation (conversion after a 1-h reaction time and selectivity to unsaturated alcohols (UA) at 30% citral conversion), binding energy (eV) of Pd 3d5/2 and Sn 3d5/2 core levels and atomic surface ratios obtained by XPS for 1 wt% Pd monometallic and 1 wt%Pd–xwt%Sn bimetallic catalysts supported on SiO2 and reduced at 300 °C.
| Catalyst sample | Pd | Pd–1 wt%Sn | Pd–1.5 wt%Sn | Pd–2 wt%Sn |
| Citral conversion (%) | 100 | 95 | 10 | 9 |
| UA selectivity (%) | 0 | 12 | 82 | 77 |
| Pd0 3d5/2 (eV) (FWHM) | 331.9 (1.80) | 331.8 (1.58) | 332.0 (1.36) | 332.1 (1.34) |
| Sn0 3d5/2 (eV) | — | 482.0 | 482.5 | 482.5 |
| Sn2+,4+ 3d5/2 (eV) | — | 485.0 | 485.2 | 485.0 |
| Sn0/Sntotal | — | 0.13 | 0.25 | 0.17 |
| Sntotal/Pd | — | 1.57 | 1.26 | 2.40 |
| Sn0/[Sn0 + Pd] | — | 0.17 | 0.25 ≈ Pd3Sn | 0.29 ≈ Pd3Sn |
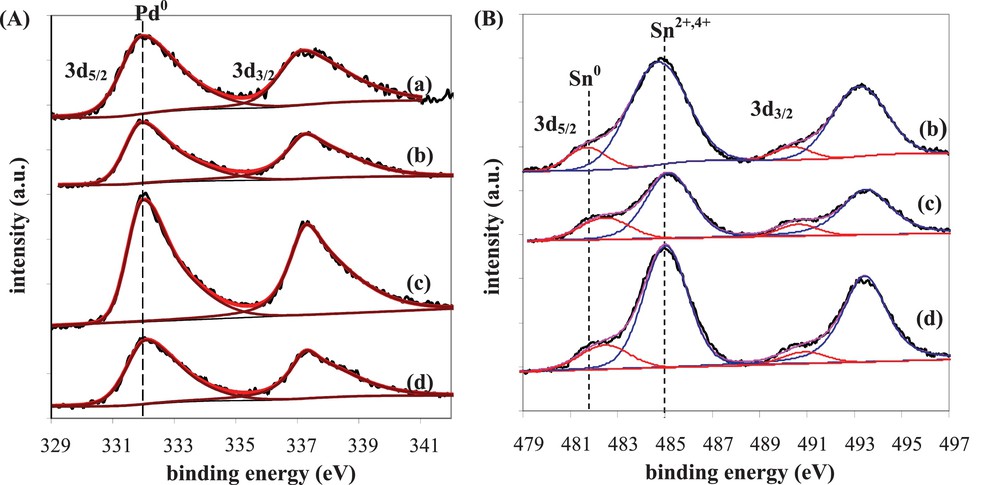
(A) Pd 3d and (B) Sn 3d spectra of Pd–Sn/SiO2 catalysts prepared by successive impregnations and reduced at 300 °C: (a) 1 wt%Pd/SiO2, (b) 1 wt%Pd–1 wt%Sn/SiO2, (c) 1 wt%Pd–1.5 wt%Sn/SiO2, (d) 1 wt%Pd–2 wt%Sn/SiO2.
4 Conclusions
Bimetallic catalysts, and notably when prepared by redox techniques, allow us to finely tune activity and selectivity for hydrogenation reactions in liquid phase. In the present paper, advantages of these preparation methods are exemplified with two reactions processes: selective hydrogenation of citral and nitrate reduction in water. The addition of a second metal allows us to improve the selectivity in citral hydrogenation to unsaturated alcohols and to improve the activity in nitrate reduction. In spite of very different reaction conditions, some rules can be put forward to explain the results:
- • an intimate contact between the two metals is strictly required to contribute to improve the catalytic performances for both reactions. The second metal may be in excess but a significant fraction should be located in the upper layer of the parent metal particles;
- • in many instances, the second metal is in an oxidized state. The specific interaction between M1 (parent metal) and M2δ+ (additive) allows one to create selective sites for citral hydrogenation towards unsaturated alcohols. In nitrate reduction, the second metal favours the first step of the reaction (conversion of nitrate to nitrite) by a redox process during which M2 is constantly in an intermediate state between metal and ionic species;
- • alloy formation between M1 and M2 is generally not required but, in few cases, alloys may improve significantly catalytic performances, as illustrated with the example of Pt–Sn/SiO2 catalysts in which the Pd3Sn phase is very selective to hydrogenate citral towards unsaturated alcohols.