

1 Introduction
A significant fraction of the carbon atoms present in dissolved natural organic materials bears carbonyl groups [1]. These carbonyls, and more particularly aromatic carbonyls, are thought to be important photooxidants for various phenols present in natural waters. Aromatic ketones absorb in the natural emission spectrum of the sun and their photoactivated states react with phenols at rates close to the diffusion-controlled limit [2]. Canonica and co-workers brought into evidence that this photooxidation involved proton or electron transfer from the triplet excited state of the carbonyl functionalities, which proved to be important oxidants despite their rather short lifetime [3,4]. Aromatic ketones, like benzophenone, also oxidize efficiently sea salt halides after absorption of actinic sunlight [5]. Photosensitized reactions of aromatic carbonyls are not only a known pathway for the formation of oxidants in natural waters, but proved also to be an important source of hydrogen peroxide in atmospheric aerosols [6].
Imidazole-2-carboxaldehyde (IC), bearing a carbonyl function, and other imidazoles get growing interest in atmospheric chemistry, since their presence in secondary organic aerosols (SOA) has been explained by an in situ catalytic process involving ammonium salts and glyoxal, an ubiquitous organic compound in the troposphere [7,8]. The formation of this catalytic product in aerosols explains part of the missing glyoxal in the gas phase through an uptake of glyoxal that was ignored for a long time [9,10]. Recently, it has been shown that imidazole-2-carboxaldehyde, one of the products formed by the ‘iminium-pathway’, can efficiently photo-induce the growth of aerosols [11]. This compound is thus a secondary formed photosensitizer and can play an important role in SOA growth in the absence of gas-phase oxidants under light conditions. The presence of these nitrogen-containing compounds in particles can also lead to changes in the optical properties of the aerosol, hence affecting the aerosols’ radiative forcing and cloud properties [12,13].
Imidazole compounds are also known to be present in the sea and in the sea-surface microlayer (SML) [14,15]. Glyoxal has been measured in the marine boundary layer (MBL) [16] and amines are an important constituent of marine aerosols [17], which comforts the hypothesis of formation of imidazoles in marine environment. The SML shows also enrichment in nitrogen during periods of high biological activity [18]. The organic-enriched SML, the thin interfacial zone between the atmosphere and the subsurface water, presents different chemical and physical features from those of the underlying water and plays an important role in the sea–air chemical interactions. Ozone loss from the marine boundary layer (MBL), for example, can occur through the reactive uptake of ozone by plant exudates such as chlorophyll present in the SML [19–21]. SML samples also feature higher absorption in the UV-region than the bulk, implying that photochemical processes can be important in transformations in and between dissolved and particulate matter [18].
Photoactive compounds, found in a higher concentration in the SML than in the underlying water, can also produce active halogen species after oxidation of the halides present in the MBL, both as aerosols and in the sea water. Photoactivated aromatic carbonyls, like benzophenone, and other photoactive compounds like chlorophyll, can lead to the formation of the reactive radical anion (X2•–) and of molecular halogens, which can be released into the gas phase [5,22]. This kind of direct photochemical reactions at the interface could account for a part of the halides observed over open oceans [23]. Gas-phase halogen atoms, especially iodine and bromine, will set in catalytic cycles of ozone destruction in the MBL, thus changing locally the oxidizing capacities. Gas-phase iodine chemistry can also give rise to particle formation [24,25], while chlorine will be inclined to oxidize hydrocarbons [26]. Other sources of such reactive halogen atoms in the MBL include photolysis of polyhalogenated compounds emitted by algae [27], heterogeneous chemistry of ozone or NOy on snow or sea salt particles [28] and, particularly for chlorine, photochemical reactions at the air–sea interface [29]. Halogen chemistry is thus profoundly entwined with aerosol production and MBL oxidizing strength, affecting potentially cloud condensation nuclei and hence climate change.
The aim of this work is to explore the photochemical properties of imidazole-2-carboxaldehyde (IC), a secondary photosensitizer potentially present in marine environment and known to induce particle growth as a secondary formed photosensitizer and to examine the bimolecular rate coefficients of this compound in the presence of halide anions, Cl–, Br– and I–, by means of laser flash photolysis.
2 Experimental
2.1 Laser flash photolysis
The transient absorption spectrum of the excited IC was followed with a classical laser flash photolysis apparatus [20]. The photolysis excitation source was the third harmonic (266 nm, pulse width ∼7 ns) of an Nd:YAG laser (Surelite II 10, Continuum) operated in the single-shot mode. The choice of the wavelength is justified by the maximum of absorption of IC at 290 nm, as can be seen from the absorption spectrum shown in Fig. 1, even if it has been shown that at wavelengths > 290 nm, IC can induce a photosensitized reaction [11].
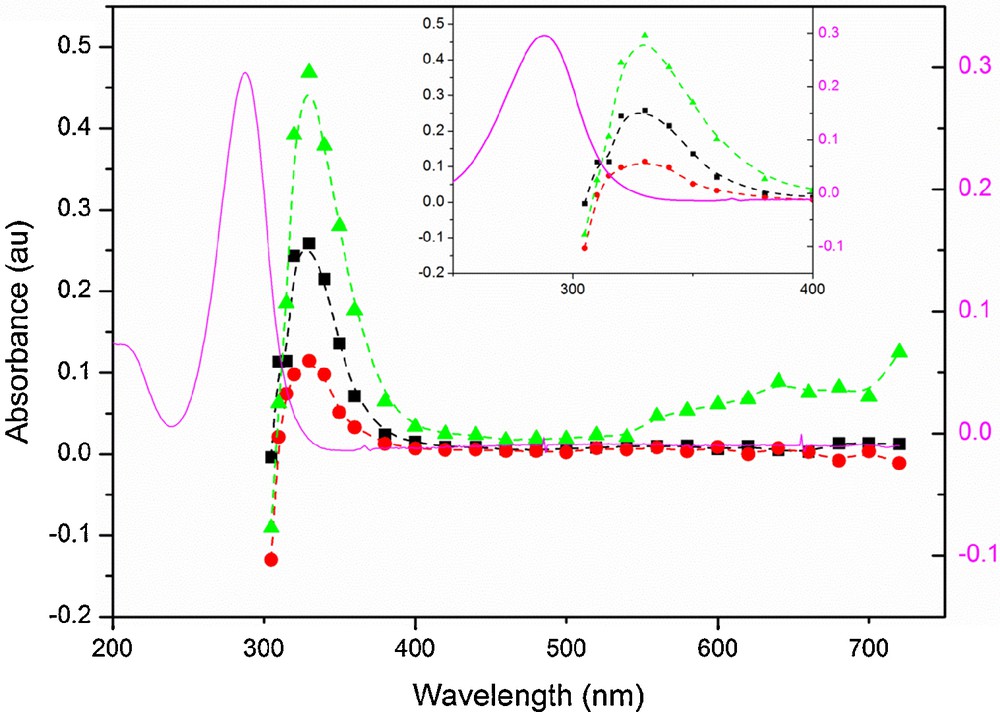
(Color online). Absorption spectrum of an aqueous solution of IC (0.25 mM) recorded 330 ns (■), 1 μs (●) and 2 μs () after the laser pulse. The solid line (–) shows the absorption spectrum of a solution of 0.25 mM IC (right Y scale). The insert is a zoom of the spectrum from 250 nm to 400 nm.
During the experiments, the laser pulse energy was limited at 15 mJ per pulse (∼39 mJ/cm2) to limit as much as possible photolysis of the photosensitizer and therefore possible interferences of its products on the studied chemistry, but also two-photon excitation. The laser output passed through the aperture in the short axis (4 mm path length) of a fully masked quartz flow cell, mounted at 13 cm of the laser output. The solution containing the photosensitizer was introduced in the flow cell of 450 μL by means of a peristaltic pump, with a flow of 1.6 mL/min, ensuring a complete purge of the exposed volume every 17 s. This limited the exposition of the introduced solution to 3–4 laser shots and maintained a constant temperature in the flow cell. All connections where made from either glass or PTFE tubing, ensuring a clean liquid flow.
Transient species produced by the pulsed laser beam were monitored by means of time-resolved absorption spectroscopy. The analyzing light, provided by a 75 W high-pressure Xenon arc lamp (LOT-Oriel), passed through the two apertures of the long axis of the flow cell (1 cm path length). The light was then collected by a ¼-m monochromator (Spectral Products DK240) equipped with a 2400 grooves/mm grating and detected by a photomultiplier (Hamamatsu H7732-01). The PMT signal was passed through a high-speed current amplifier/discriminator (Femto) and the AC component recorded on a 300 MHz oscilloscope (Tektronix TDS3032c). The digitalized signal was then transferred to a computer for further processing. Typically, 16 measurements were averaged at each wavelength to obtain a transient absorption spectrum, with an accuracy of 3%. The full transient absorption spectrum was then reconstructed from the steady and transient signals. For construction of an absorption spectrum, measurements were repeated every 10–20 nm between 320 nm and 720 nm, limits determined by the measuring system; for the kinetics, the probe wavelengths for the transient absorption decay of triplet state of IC were 330 nm and 350 nm.
For the analysis of triplet state decays of IC in pure water, linear regression on a logarithmic plot of the data and a mono-exponential decay model fits were performed on the same datasets. As the results of both fitting methods were comparable, we will here only present the results obtained by non-linear fits.
2.2 Materials and solutions
All the experiments were performed at room temperature in the range 295–300 K. Aqueous solutions containing 0.25 mM of IC were deoxygenated with nitrogen for 20 min prior to analysis and kept under a continuous stream of nitrogen during the experiment. All solutions were freshly prepared using 18-MΩ ultra pure water (ELGA) and were put in an ultrasonic bath for 10 min to promote solubility. The chemicals used in this study–imidazole-2-carboxaldehyde (Aldrich, 97%), sodium chloride (> 99% Sigma-Aldrich), sodium iodide (> 99% Sigma-Aldrich), sodium bromide (> 99% Sigma-Aldrich)–were all used without further purification. The known impurity of bi-imidazole in the commercial imidazole-2-carboxaldehyde did not notably interfere, since no absorption for triplet-state bi-imidazole was detected. In our conditions, i.e. in aqueous solutions of pH 6.5, IC is expected to be under its aldehyde form [30].
3 Results
3.1 Characteristics of the triplet state imidazole-2-carboxaldehyde
Laser flash photolysis of N2-saturated solutions of 0.25 mM IC in pure water showed strong transient absorptions, as shown in Fig. 2. The transient absorption of excited IC also shows intense fluorescence in the first nanoseconds after the laser pulse. Fig. 1 shows recorded transient absorption spectra at different time intervals after the laser pulse, demonstrating the uniform decrease of the absorbance without formation of any significant band. The absorption spectrum can thus be attributed to the triplet–triplet absorption of 3IC* and was recorded for the first time, since very little is known about the photochemistry of IC. The transient absorption spectrum recorded after the laser pulse is characterized by a strong absorption peak with a maximum absorption wavelength around 330 nm and a weaker broad absorption band around 650 nm. For the determination of lifetimes and quenching rate coefficients, the decay of the triplet state was monitored at 330 nm.
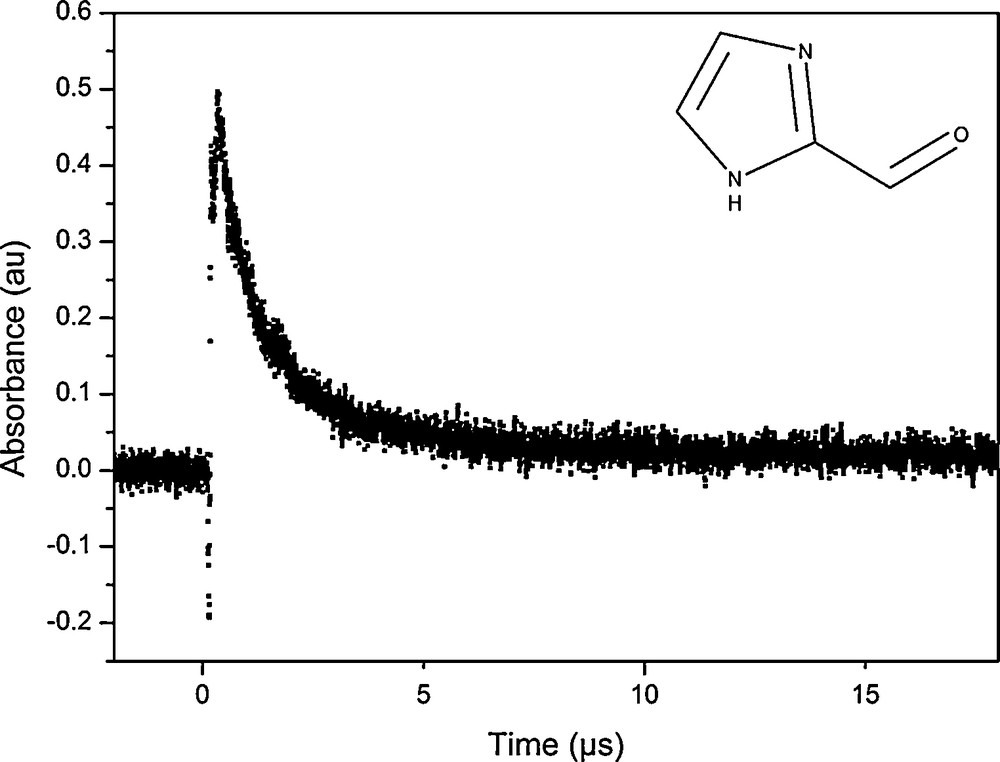
Transient absorption decay obtained at 330 nm of the IC triplet state in a deoxygenated aqueous solution of 0.25 mM IC.
The absorption decay trace of excited 3IC* at 330 nm fitted well a single exponential process of the form:
| (1) |
| (2) |
In the presence of oxygen, the lifetime of the triplet state decreased in an oxygen-saturated solution, which indicates that an energy transfer to oxygen is possible. In an air-saturated solution, the quenching constant for the reaction with oxygen was estimated at 3.9 × 109 M−1s−1, assuming that the concentration of oxygen is described by Henry's law. This fast energy transfer could generate reactive oxygen species such as singlet oxygen or superoxide anions.
3.2 Quenching
The reaction of 3IC* with three sea salt halides i.e. chloride, bromide and iodide, was investigated. These salts are present in different and varying concentrations in seawater (and aerosols); the average chloride concentration is around 0.54 M, bromide concentration around 0.84 mM, and iodide varying between 0.02 μM and 0.4 μM [33,34]. Previous studies have shown that sea salts, like phenols, can act as electron donors towards the excited carbonyls forming reactive halide radicals [4,5]. The reaction mechanism for the growth of aerosols containing traces of IC, proposed in Aregahegn et al. [11], suggests that the excited state 3IC* can act through a proton or an electron transfer from limonene towards IC. In this study, photoactivated IC is expected to interact with the halide anions by electron transfer to produce atomic halogens [5]:
| 3IC* + X– → IC•– + X• | (E.1) |
The reactivity of the three quenchers was investigated by introducing NaI, NaBr or NaCl in different concentrations in deoxygenated aqueous solutions of IC, with a large excess of the quencher compared to the photosensitizer, respecting pseudo-first-order conditions. The quenching rates coefficients for 3IC* in the presence of halides were determined by the Stern–Volmer equation (eq. (3)):
| (3) |
3.2.1 Iodide
The decay of the triplet–triplet absorption monitored at 330 nm in the presence of NaI showed still a mono-exponential decay, but decreased much faster in the presence of an increasing concentration of I– (Fig. 3).
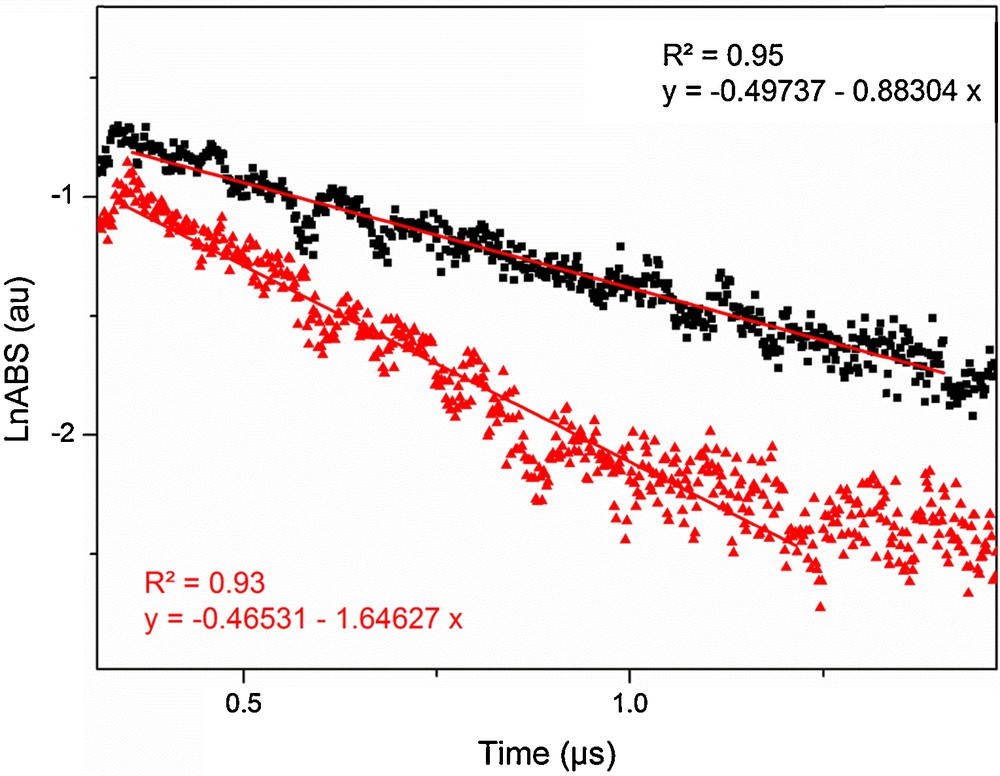
(Color online). Log plot of a transient absorption decay obtained at 330 nm of the IC triplet state in a deoxygenated aqueous solution of 0.25 mM IC alone (■) and in the presence of 0.25 mM NaI (); the linear fits show a larger slope in the presence of NaI.
The quenching rate coefficient kq(I–) obtained for 3IC* by I– from the slope of the Stern–Volmer plot shown in Fig. 4 is kq(I–) = (5.33 ± 0.25) × 109 M−1 s−1. The high rate coefficient obtained is close to the diffusion-controlled limit and shows that quenching by iodide anion is highly efficient.
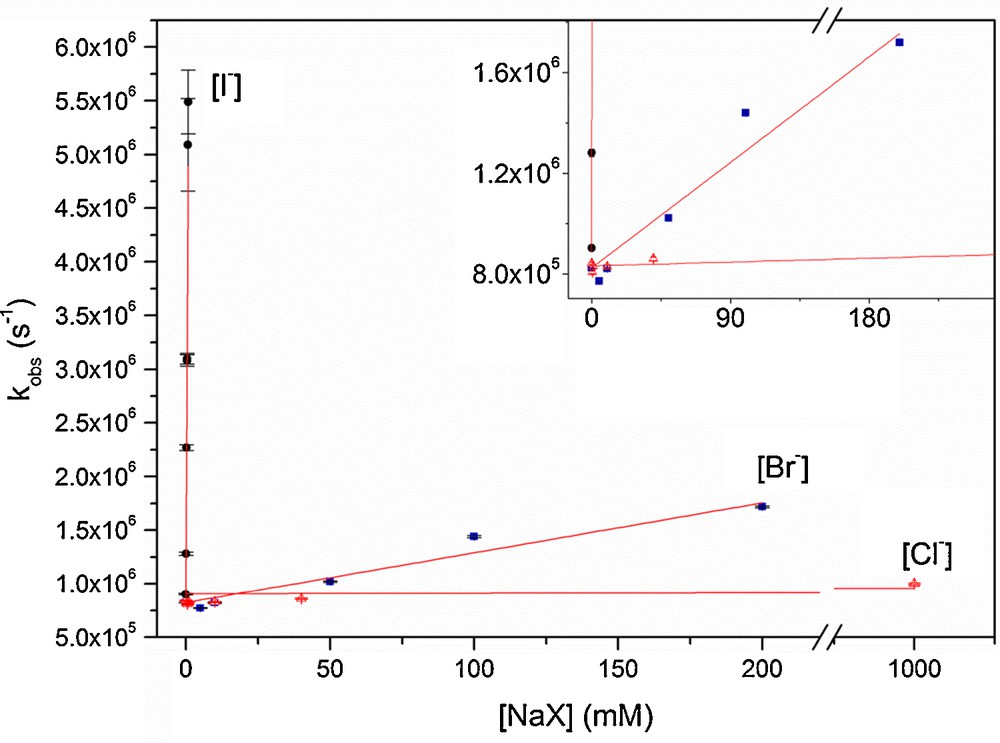
(Color online). Stern–Volmer plot of the pseudo-first-order rate coefficients kobs of IC triplet state quenching as a function of halide concentrations.
3.2.2 Bromide
Triplet–triplet absorption decay of IC was also faster in the presence of increasing concentrations of NaBr, remaining mono-exponential at 330 nm for concentrations of bromide < 200 mM. From the Stern–Volmer plot (Fig. 4), the quenching rate coefficient determined was kq(Br–) = (6.27 ± 0.53) × 106 M−1 s−1 for bromide. The quenching rate coefficient is orders of magnitude lower than the quenching rate for iodide and also quite slow compared to the rates obtained with other photosensitizers, such as benzophenone, where values for quenching rates for bromide are between 2.2 and 3.5 × 108 M−1 s−1 [5,33].
3.2.3 Chloride
In the presence of chloride, the absorbance of 3IC* showed only little-decreased lifetimes, even with chloride concentrations up to 1 M (Fig. 4). The quenching rate coefficient determined is kq(Cl–) = (1.31 ± 0.16) × 105 M−1 s−1 and in view of the coefficients obtained for the other halides, we can conclude that the quenching of triplet state IC by chloride is not efficient.
3.2.4 Comparison of the three quenchers
When considering the three quenching rate coefficients, it comes into evidence that the quenching rate coefficients are linked to the oxidation potentials of the used halides. Table 1 lists the quenching rate coefficients and the oxidation potentials for each halide. For the halide with the lowest oxidation potential, iodide, the quenching rate coefficient obtained is four orders of magnitude higher than the quenching rate coefficient obtained for chloride. It seems thus probable that the excited triplet state of IC is quenched by these halides by an electron transfer towards 3IC*. Electron transfer from a halide (X–) to the excited triplet state of a carbonyl compound yields a halide radical and a ketyl radical.
The oxidation potential E(X–/X) of each halide and their quenching rate coefficients kq obtained for 3IC*.
| Quencher | E (X/X–) (V) [35] | kq (M−1 s−1) |
| Cl– | 2.6 | (1.31 ± 0.19) × 105 |
| Br– | 1.93 | (6.27 ± 0.53) × 106 |
| I– | 1.33 | (5.33 ± 0.25) × 109 |
Such an intramolecular electron transfer is usually described by a mechanism, schematized in equation (E.2), involving the formation of a contact complex, after which the electron transfer takes place and a charge complex is formed. Finally, the charge complex breaks up and oxidized donor and reduced acceptor are separated:
| 3(IC)* + X– ⇋ [3(IC)* ••• X–] ⇋ [IC•–••• X•] → IC•– + X• | (E.2) |
This mechanism requires the free enthalpy of the electron transfer (ΔGET) to be negative or close to zero for electron transfer to become thermodynamically favorable [5,36]. It could be interesting to compare the free enthalpy of the electron transfer for each quencher, if the reduction potential of IC and the energy of the triplet state of the photosensitizer were known. It seems that iodide, the halide with the lowest electronegativity, will be the best electron donor and consequently the most efficient quencher, hence the highest rate coefficient determined. Chloride on the other hand displays the highest electronegativity and oxidation potential and is a poor electron donor. The quenching rate coefficient was, as expected, lower for chloride than for bromide and iodide. The energy of triplet state IC is thus expected to be higher than the oxidation energy of iodide and lower than the oxidation energy of bromide, since the rate for the electron transfer from iodide is close to the diffusion-limited rate, but substantially lower for bromide. The electron transfer from chloride is thermodynamically unfavorable, as is reflected by the very low rate coefficient.
4 Discussion
In mechanism (E.2) proposed above, halogen atom radicals are formed. These radicals are very reactive, can form secondary products and give rise to transient product absorption. By establishing transient absorption spectra at different times after the laser pulse and for different concentrations of halogens, transient absorption products could be detected in the presence of iodide and bromide. No radical product formation was observed following the excitation of IC in the presence of chloride, since the quenching reaction with chloride was shown to be much slower.
4.1 Iodide
As mentioned in the previous section, the decays of absorption of 3IC* in the presence of iodide could be fitted by a mono-exponential function at wavelengths around 330 nm, the maximum absorption of 3IC*. At longer wavelengths, particularly at 380 nm, as illustrated by Fig. 5, the decays fitted a bi-exponential decay of the form:
| (4) |

(Color online). Transient absorption decay of 3IC* (a) in the presence of 300 mM NaBr (■) monitored at 355 nm and (b) in the presence of 0.4 mM NaI (Δ) monitored at 380 nm, showing a bi-exponential decay.
After comparison of absorption spectra at different times after the laser pulse, the absorption at 380 nm of the species with the longer lifetime could be observed several microseconds after the laser pulse and was stronger, though still low, than that of a solution of IC in pure water (Fig. 6). The comparison of the absorption spectrum of 3IC* in pure water with the absorption spectrum of 3IC* in the presence of 0.4 mM of NaI after shorter time intervals after the laser pulse, illustrated by the insert of Fig. 6, does not feature this enhanced absorption around 380 nm. This indicates clearly a secondary formation for the species absorbing at 380 nm.

(Color online). Absorption spectrum of IC (0.25 mM) in the presence of NaI (0.4 mM) registered 8 μs after the laser pulse, showed as ---Δ, featuring a peak around 385 nm, not observed in the absorption spectrum of IC alone (■). At shorter times after the laser pulse (700 ns, insert), the spectrum of IC in the presence of NaI (---Δ) is less intense at all wavelengths than that of IC only (■).
One of the products that are possibly formed by the atom radical I• is the radical anion I2•–, formed by the combination of the anion with the oxidized halide radical. This well-known transient species is relatively stable with absorption maxima at 385 nm and 725 nm, hence showing a long lifetime [37–39]. Our system did not permit to detect the second absorption peak of I2•– at 725 nm, whose intensity is expected to be 3.5 times lower than that of the peak around 385 nm. The peak of I2•– is low, despite the rather high molar absorption of this transient species (9600 cm−1 mol−1) [38]. This could be explained by rapid I2 formation [40] or by fast scavenging reactions of I• and I2•–, limiting the amount of I2•– detected, as has been showed in the case of chloride [41]. These radical reactions are possible with the solvent or with the photosensitizer [5,42], which could explain the high absorption, even 8 μs after the laser pulse at wavelengths < 330 nm (Fig. 6). The radical I2•– is the only species expected to absorb at 380 nm and the peak can thus be distinctively attributed to this transient species. The absorption at shorter wavelengths cannot be completely monitored with our system and would not be characteristic of one expected transient species.
4.2 Bromide
The transient species formed after the photooxidation of bromide by 3IC* were more difficult to follow, since the absorbance of one the expected products, Br2•–, was overlapping with the absorbance of triplet state IC. Nonetheless, the fitted curves at the maximum absorption, 330 nm, of the triplet state IC showed a mono-exponential decay in the presence of bromide concentrations in the range from 1 to 200 mM. The decay became bi-exponential for wavelengths around 354 nm, the maximum absorption of Br2•– in water [43], indicating secondary product formation. As in the case of iodide, two species with distinct lifetimes could be distinguished, the short-lived ascribed to the decay of 3IC*. On the absorption spectrum (Fig. 7) taken 10 μs after the laser pulse, the absorption around 360 nm is higher in the presence of bromide than that obtained with IC alone. This could be due to the formation of Br2–•, a strong absorbing radical species (ɛmax = 9900 M−1 cm−1 [40]) formed from the reaction of Br• with Br–.
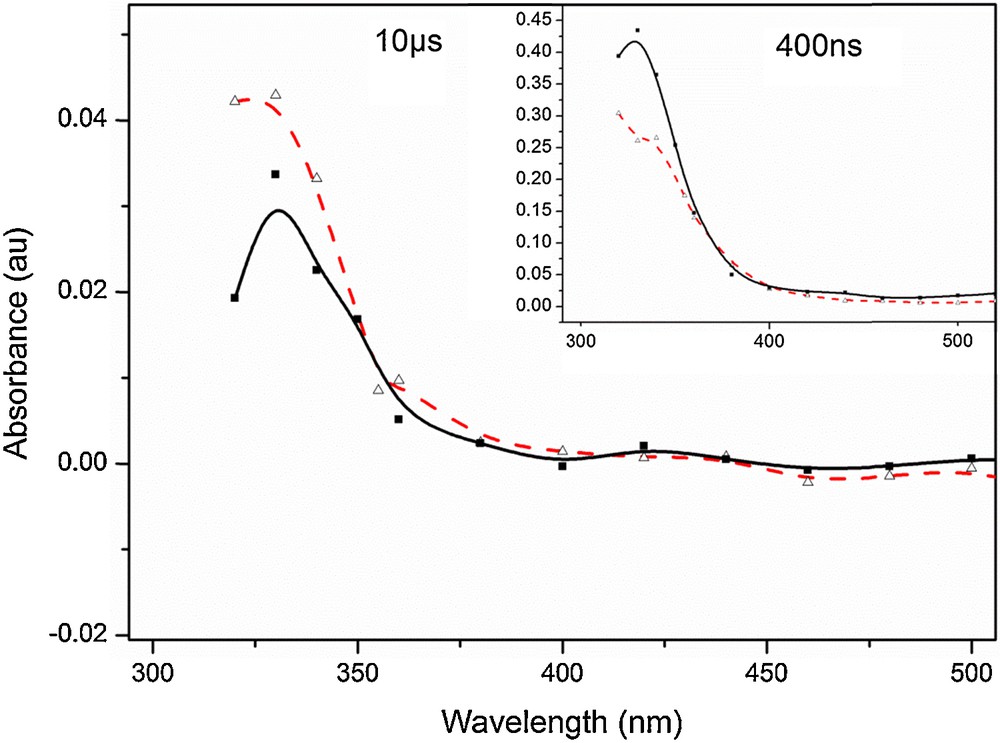
(Color online). Absorption spectrum of IC (0.25 mM) in the presence of NaBr (300 mM) showing higher absorption around 360 nm (---Δ) than the absorption spectrum of IC alone (■), registered 10 μs after the laser impulse. At shorter times after the laser impulse (400 ns, insert), the spectrum of IC in the presence of NaBr (---Δ) is less intense at all wavelengths than that of IC only (■).
Fig. 7 also shows increased absorption for wavelengths shorter than 355 nm, even at long times after the laser pulse, absorption not explained by the triplet state IC absorption. This absorption, detected only partially with our apparatus, shows a large peak with a rather long lifetime. Products contributing to this absorption could be recombination products of IC with Br• or the very strong absorbing Br3– (ɛmax = 40,900 M−1 cm−1, λmax = 266 nm [44]).
The absorption of Br2•– seems rather low, indicating that Br2•– is not a major product of this photooxidative process. This could be due to Br2 formation or scavenging reactions of Br• and Br2•– by the solvent or the photosensitizer [45]. One of the products possibly formed from the reaction of Br• with water is BrOH•– [45]. This radical shows a maximum absorption at 352 nm, very similar to, but less intense than the absorption of Br2•– [43]; nevertheless, the reaction of Br• with water is very slow (k = 1.4 s−1, in [45]). Even if the absorption at this wavelength cannot be unambiguously ascribed to Br2•–, the formation of BrOH•– can thus be considered as negligible.
4.3 Proposed mechanism
To resume the reactions and the formation of the products detected, the proposed mechanism involves the following reactions:
| IC* + X– → IC•– + X• | (R.1) |
| X• + IC → products | (R.2) |
| X• + X– ⇋ X2•– | (R.3) |
| X2•– + IC → products | (R.4) |
| X2•– + X2•– ⇋ X3– + X– | (R.5) |
| X3– ⇄ X2 + X– | (R.6) |
| X• + X• → X2 | (R.7) |
| X2•– + X2•– → X2 + 2X– | (R.8) |
| X2 ⇄ X2 (g) | (R.9) |
This mechanism explains the formation of reactive radicals and can lead to the formation of halogen molecules in the gas phase.
5 Summary and atmospheric implications
Our results confirm the role of IC as a photosensitizer proposed by Aregahegn et al. [11], for the first time evidencing the existence of a triplet state for this compound. The lifetime of 3IC* is estimated at 1.21 μs in a deoxygenated aqueous solution and the maximum absorbance was measured at 330 nm.
The triplet state of IC was efficiently quenched by iodide, kq(I–) = (5.33 ± 0.25) × 109 M−1 s−1, and much less by bromide, kq(Br–) = (6.27 ± 0.53) × 106 M−1 s−1. Chloride showed a slow quenching rate, kq(Cl–) = (1.31 ± 0.16) × 105 M−1 s−1 and reactions with chloride might thus be negligible. Reactions with these halides form atomic halogens, some of which could be detected by increased transient absorption. Also, the quenching rate constants show the same trends as the oxidation potential of each sea salt anion, suggesting that electron transfer is the mechanism behind the photooxidation of the halides.
Imidazole-2-carboxaldehyde proved to be a quite efficient photosensitizer and, as IC can also be formed in aerosols in the presence of glyoxal and ammonia, its presence, even in traces, could have an important influence on the particle phase chemistry at the interface of aerosols. Its photosensitized reactions can lead to an uptake of gas-phase VOCs and lead to particle growth, as demonstrated by the works in [11]. The photochemical radical production mechanism initiated by carbonyl compounds proposed in this study is potentially important in the SML, not only in reactions with sea salts, but also for the oxidation of other organic compounds with a one-electron oxidation potential. Indirectly, photochemistry evidenced for IC and other carbonyl compounds may also influence the chemistry of the SML by the radicals and secondary products formed during this process and the MBL by the release of reactive (halogenated) species, such as Br2, Br2•–, I2 and I2•– in the gas phase [5,11].