

1 Introduction
In the field of liquid-phase chemical hydrogen storage, aqueous alkaline solutions of sodium borohydride (sodium tetrahydroborate, NaBH4) have shown to be attractive, especially for low-temperature fuel-cell applications. The solution can be used “directly” as a liquid fuel of a direct borohydride fuel cell that is a technology based on the anodic electro-oxidation of the borohydride anions BH4− [1]:
| BH4− (aq) + 8 OH− (aq) → BO2− (aq) + 8 e− + 6 H2O (l) | (1) |
Otherwise, the solution can be used as the “indirect fuel” of the fuel cell, in fact as a hydrogen carrier for fuel cell [2]. In this case, the couple NaBH4–H2O is dehydrogenated by hydrolysis (Eq. (2)) in the presence of a metal-based catalyst [3]; the as-generated hydrogen is then oxidized at the anode of a fuel cell [2]:
| BH4− (aq) + 4 H2O (l) → B(OH)4− (aq) + 4 H2 (g) | (2) |
The advantageous features of this reaction are as follows. First, the gravimetric hydrogen density of NaBH4·H2O (Eq. (2)) is 7.3 wt% H, which is a high capacity if compared to solid- and liquid-state hydrogen storage materials taken in near-ambient conditions [4]. Higher densities (up to 9 wt% H) can be achieved at higher temperatures (> 80 °C) when solid sodium borohydride and steam are used [5]. Second, the reaction is spontaneous, thus exothermic, with ΔH between −210 and −250 kJ mol−1 [6]. To avoid uncontrolled generation of hydrogen, the solution is generally stabilized by adding some sodium hydroxide NaOH (e.g., 0.1–5 M) into it [7]. With such a stabilized aqueous solution, dehydrogenation of sodium borohydride by hydrolysis requires then a catalyst [8]. Third, the liberated hydrogen is pure [9].
The development of the hydrolysis of aqueous alkaline solutions of sodium borohydride is however hindered by an important issue, which is related to the formation of the tetrahydroxyborate anion B(OH)4−. Indeed, it is highly crucial to recycle this by-product back into the starting material, i.e. BH4−. The hydrogen cycle with sodium borohydride must be closed [10]. Yet, the strength/energy of the B − O bond in B(OH)4− is comparable to that of the CO bond in CO2 and, therefore, the conversion of B(OH)4− into BH4− is thermodynamically difficult [11]. Actually, the inefficiency and the high cost needed for such a process have made the US Department of Energy (US DOE) recommend, in 2007, a no-go for sodium borohydride for on-board automotive applications [12]. Another reason for this decision has been motivated by the low effective storage capacities achieved [13]. Despite everything, aqueous alkaline solutions of sodium borohydride are considered to be attractive for mobile/portable applications, e.g. niche applications [14].
During the past 13 years dedicated to hydrolysis of sodium borohydride, priority has been given to searching for active metal-based catalysts [3], and especially cobalt-based catalysts [15]: e.g., cobalt halides that lead to in situ formation of aggregated nanoparticles when put into contact with sodium borohydride (a strong reducing agent) [16]; ex situ-prepared cobalt nanoparticles and nanostructures [17]; cobalt deposited on powdery supports, like, e.g., alumina Al2O3, titanium oxide TiO2 or even clay [18]; cobalt deposited on shaped substrates like, e.g., nickel foam, polycarbonate membrane or copper foil [19]; cobalt alloyed with other elements like, e.g., phosphorus, molybdenum or chromium [20]; and so on. For more details, the reader is invited to refer to the reviews cited in reference [3]. It is worth noting that despite the huge work dedicated to cobalt in hydrolysis of sodium borohydride, the catalytically active form of the metal that is really involved in the reaction is rather unknown [15].
From a fundamental point of view, one can claim that knowledge of the hydrolysis mechanisms, especially those occurring on the surface of cobalt catalysts, is quite poor. In our opinion, this is due to three main reasons [15]. The first one is related to the difficulty to perform in situ and operando characterizations, which are essential to follow any evolution of the cobalt catalyst surface at the molecular level during the reaction. Actually, vigorous bubbling of hydrogen and precipitation of borates make any characterization very difficult. The second reason is related to the (very) fast kinetics of hydrolysis, which is generally concomitant with an important temperature increase (due to ΔH between −210 and −250 kJ mol−1 [6]). The third reason is that the nature of the catalytically active phase of cobalt is still unknown. Cobalt–boron alloys, cobalt borides, metal cobalt, cobalt oxides and so on have been suggested up to now. Recently, we have demonstrated that the catalytic surface of cobalt is not stable and is modified in the course of the hydrolysis reaction, leading to the deactivation of the catalyst [21]. This is a further complication. We have shown that the surface of fresh cobalt supported on nickel foam is initially clean (i.e. free of surface-adsorbed species), but after hydrolysis, the surface is covered by a film consisting of strongly-adsorbed (poly)borates. This is consistent with Kim et al.’s observation about nickel filamentary catalysts [22]. Consequently, we have suggested that the catalytically active phase of cobalt is a core@shell-type structure where the core is cobalt and the shell (poly)borates (Fig. 1).
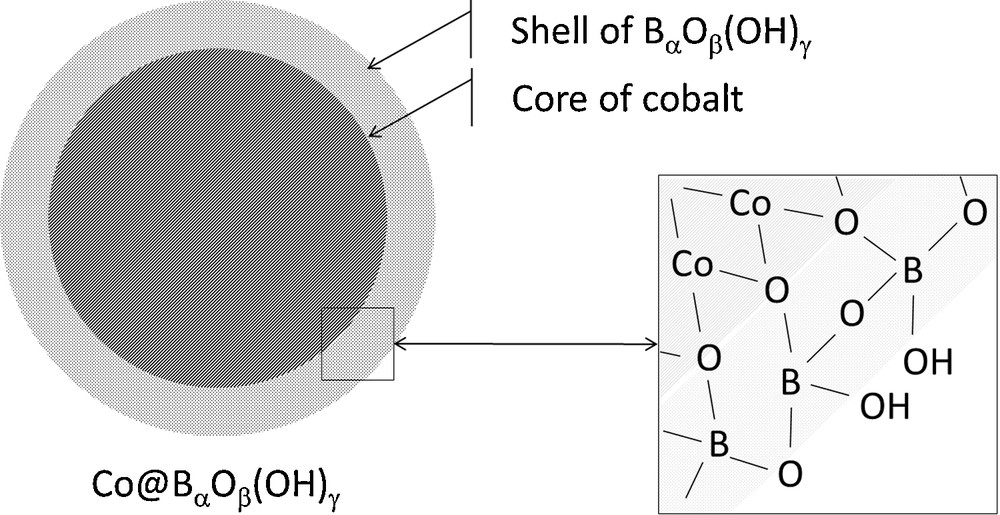
Schematic representation of Co@BαOβ(OH)γ, a core@shell-type structure where the core is a cobalt compound and the shell (poly)borates.
Adapted from [15,21].
Despite the aforementioned difficulties, which can be simply explained in terms of experimental problems, mechanistic schemes have been tentatively elaborated and proposed in the open literature. In some of those works, kinetic studies based on known kinetic models (e.g., Langmuir–Hinshelwood and Michaelis–Menten models) were conducted in order to gain new insight into hydrolysis mechanisms. These studies are reported and discussed in details herein. In this context, we propose a short review aiming at (i) reporting the hydrolysis mechanisms proposed up to now as well as the kinetic models used for this purpose, and (ii) discussing their reliability on the basis of the original phase of the catalytic cobalt (core@shell-type structure) we found.
2 Kinetic studies and models
There are various kinetic models that can be used to fit the experimental data (generally the volume or mole number of hydrogen as a function of time) of metal-catalyzed hydrolysis of aqueous alkaline solution of sodium borohydride and then to give a first interpretation of the reaction mechanisms. To date, five different models have been considered: the nth-order kinetics, where n is equal to 0, 1 or 2; the power law; the Langmuir–Hinshelwood model; the Michaelis–Menten one; and the semi-empirical kinetics. These models were surveyed in 2011 by Retnamma et al. [23], who especially showed discrepancies in the determined kinetic parameters (reaction orders versus the concentration of NaBH4, versus the concentration of the stabilizer NaOH, and/or versus the catalyst amount; apparent activation energy). For more details, the reader is invited to refer to this reference. However, a short description and an analysis of the various models (in the next section) may be useful to better understand the reaction mechanisms we survey.
2.1 Power law and reaction orders
The hydrolysis of an aqueous alkaline solution of sodium borohydride involves the borohydride anion BH4− and up to four water molecules (Eq. (2)). The reaction rate can be written as a power law where the rate r is proportional to the concentration of BH4− and to that of H2O (Eq. (3)):
| (3) |
In excess of water (e.g., mole ratio H2O/BH4− >> 50), the term
| (4) |
The effect of the increase of the catalyst amount on the reaction rate will not be discussed in details hereafter, as it is generally expected that an increase means more active sites and thus an improvement of kinetics. The reaction order δ should be positive [24].
Reaction orders α versus the concentration of BH4− of 0, 1 and 2 were reported [25–28], implying zero-order, first-order, and second-order kinetics. Zero-order kinetics is generally observed for specific experimental conditions [23] since transition from zero-order to non-zero-order has been observed concomitantly with an increase in the solution's viscosity [8], solution pH [27], and/or reaction temperature [29]. For example, Patel et al. [30] reported a first-order kinetics for 0.005–0.05 mol NaBH4 L−1 and a zero-order one for higher concentrations (up to 0.25 mol NaBH4 L−1). For zero-order kinetics, the reaction rate and the catalytic activity do not depend on the concentration of BH4−, suggesting that the amount of BH4− adsorbed on the catalyst's surface is constant and the surface is saturated with adsorbed species. The rate-determining step should be the hydrolysis reaction. Retnamma et al. [23] showed that most of the works published within the decade 2000–2010 found zero-order kinetics for cobalt-, nickel- and noble-metal-based catalysts. First- and second-order kinetics were reported in few papers where noble metals were used as catalysts. For example, Patel et al. [26] investigated a carbon-supported palladium catalyst and found an order of 1 for ratios [NaBH4]/[catalyst] lower than 1. Reaction orders of 1 and 2 suggest that the catalyst surface is not totally occupied by adsorbed species. The rate-determining step should be the diffusion/adsorption of BH4− on the surface.
Water in the hydrolysis of an aqueous alkaline solution of sodium borohydride plays two roles. It is one of the reactants and, when used in excess, it is the solvent. It is therefore somehow difficult to carry out kinetic experiments in order to determine the reaction order β versus water. Yet, Zhang et al. [31] found an order of 0.68 for a Ni catalyst in the following experimental conditions: 0.2–1.9 mol NaOH L−1, 1.39–5.5 mol NaBH4 L−1, and 0–30 °C. The rate given by Eq. (5) was then proposed:
| (5) |
The positive effect of the water amount was also verified in our experimental conditions (results not published). A positive order was also shown for the hydrolysis of an aqueous solution of ammoniaborane NH3BH3, another attractive material for hydrogen generation by hydrolysis [32]. It should be mentioned that the power law in Eq. (5) shows a negative order versus BH4−. To explain such a value, it was suggested that sodium hydroxide might affect the desorption of the borates from the catalyst surface, thus affecting the catalytic site renewal rate. Accordingly, the rate-determining step should be by-products desorption, and accordingly adsorption of BH4−. Negative orders versus BH4− were also reported for carbon-supported ruthenium [33], modified carbon-supported cobalt [34], carbon aerogel-supported cobalt nanoparticles [35]. The decrease of the hydrogen generation rate with the increase of the sodium borohydride concentration may be due to enhanced alkalinity and viscosity of the aqueous solution [27,34,36].
Aqueous solutions of sodium borohydride are generally stabilized by adding 0.04 to 15 wt% of sodium hydroxide [7]. Negative orders versus the concentration of OH− are mainly found with noble metal-based catalysts [23,28,37], whereas slightly positive orders are generally obtained with non-noble metals (i.e. nickel- or cobalt-based alloys) [31,38]. This difference may be ascribed to a difference in the surface state of the catalysts. This is discussed in details in the section following the next one. However, for a cobalt-based catalyst, Liu et al. [39] observed an increase and then a decrease in the hydrogen generation rate with the increase of the concentration of OH− from 1 to 15 wt%, with the optimum being found for 5 wt%. The authors conjectured that the anions OH− would compete with the transfer of BH4− to the catalyst surface. Nie et al. [40] reported similar trends for Ni–Fe–B catalysts.
2.2 Bimolecular kinetic models
An alternate approach to fit the experimental data of hydrolysis of aqueous alkaline solution of sodium borohydride is to use known kinetic models. The Langmuir–Hinshelwood model applies to bimolecular reactions (Fig. 2). It assumes that, first, both reactants adsorb on free surface sites and then a reaction between these species occurs [41]. The rate expression for this model is as follows [42]:
| (6) |

(Color online). Schematic representation of the Langmuir–Hinshelwood, Michaelis–Menten, and Eley–Rideal models, involving two reactants (R1 and R2), adsorbing onto two sites of different natures (S1 and S2) or onto one site (S), and leading to the formation of two products (P1 and P2).
In this equation, k and Ka are the rate constant and the adsorption constant, respectively. The reaction order should be nil and equal to 1 at high and low concentrations of BH4−, respectively. Zhang et al. [31] analyzed their experimental data (obtained with a carbon-supported ruthenium catalyst used in a solution at 0.8 wt% NaBH4 and 3 wt% NaOH) on the basis of this model. It was assumed a two-step reaction, with (i) the equilibrated adsorption of BH4− and (ii) the reaction of the adsorbed BH4− with adsorbed H2O. Similarly, Hung et al. [42] showed a reasonable behavior description for an alumina-supported ruthenium catalyst over the entire time span of the experiment performed and within the temperature range 10–60 °C. Hence, the model is able to describe simultaneously the zero-order kinetics occurring at low temperatures and low conversions, and the first-order kinetics found at higher temperatures. For Andrieux et al. [43], the Langmuir–Hinshelwood model satisfactorily captured the behavior of cobalt nanoparticles at 10–90 °C and for a concentrated solution of NaBH4 (ca. 19 wt% or 6.2 mol L−1). More recently, Sousa et al. [44] arrived at similar conclusions for a nickel-foam-supported ruthenium catalyst.
Another kinetic model to capture the experimental data of the hydrolysis of an aqueous alkaline solution of sodium borohydride is the Michaelis–Menten one, which is generally used in enzyme kinetics. This is also a bimolecular mechanism (Fig. 2), but it assumes the reversible adsorption of only one reactant, in the present case BH4−, on the catalyst surface (with [M]0 the concentration of the surface active sites). An intermediate complex MBH4 forms and reacts with four water molecules (not adsorbed) to produce B(OH)4−, which desorbs, and 4 H2. De facto, the catalyst surface is regenerated. As reported by Dai et al. [45], the rate expression for this model is:
| (7) |
There is a last kinetic model that could be envisaged for interpreting the kinetic data of the hydrolysis of aqueous alkaline solutions of sodium borohydride. This is the Eley–Rideal model [46]. It has never been considered for the reaction discussed herein. This model is also based on the occurrence of a bimolecular reaction (Fig. 2). It assumes the adsorption of one of the reactants (i.e. BH4−) on a free catalytic site and then the direct reaction between adsorbed BH4− (denoted MBH4) and non-adsorbed H2O. These elementary steps are similar to those of the Michaelis–Menten model: i.e. reversible adsorption of BH4− on one free site of the catalyst (rate constants k1 and k−1), and reaction with 4 H2O molecules (rate constant k2; this is the rate-determining step) with formation of H2 and concomitant desorption of B(OH)4−. For the Eley–Rideal model, the rate expression involves the participation of both reactants as follows:
| (8) |
The steady-state approximation may be applied to [MBH4]. This means that the concentration of MBH4 is constant, which gives the equation:
| (9) |
Hence, by combining Eq. (8) and Eq. (9), and by posing
| (10) |
If one assumes a constant amount of H2O, Eq. (10) becomes similar to Eq. (7), and the Eley–Rideal mechanism can be seen as being equivalent to the Michaelis–Menten one. At low concentrations of BH4−, the reaction order should be 1. At high concentrations, the reaction order should be nil. In both cases, the reaction orders versus catalyst amount and water content are 1.
To sum up, the Langmuir–Hinshelwood and Michaelis–Menten mechanisms have shown to be acceptable to capture the kinetics of the hydrolysis of an aqueous alkaline solutions of sodium borohydride. Another model, the Eley–Rideal one, should also give similar results, given besides that in specific conditions this model is equivalent to the Michaelis–Menten mechanism. In other words, each of these models allows describing, qualitatively and to a certain extent, the reaction mechanisms [23], though they do not capture negative orders versus the concentration of BH4−. More sophisticated models, which might involve the occurrence of reaction intermediates like BH3(OH)−, BH2(OH)2−, and BH(OH)3− [47], would be required to exactly predict the experimental results whatever the conditions in terms of concentrations of sodium borohydride and sodium hydroxide, water content, and catalyst nature. However, the proposition of sophisticated models is strongly dependent on better understanding and thus exactly defining the reaction mechanisms.
3 Mechanisms of hydrolysis of NaBH4
Below is reported an overview of the scarce literature about the reaction mechanisms for metal-catalyzed hydrolysis of aqueous solution of sodium borohydride. The reader is invited to refer to the reference [23] for details about the mechanisms proposed for spontaneous and acid-catalyzed reactions.
3.1 Langmuir-Hinshelwood mechanism
One of the first mechanisms was proposed in 2002. Kojima et al. [48] reported their view of hydrolysis of BH4− in the presence of a LiCoO2-supported Pt catalyst. Consistently with the Langmuir–Hinshelwood model, they suggested that BH4− adsorbs on Pt while H2O adsorbs on the oxide support. Then, the adsorbed species interact, and the hydrides of BH4− and the protons of 2 H2O react to give 4 equiv of H2, while the oxygen atoms of H2O bind to B of BH4−. Kojima et al. proposed the formation of BO2− as the by-product. However, as demonstrated elsewhere [49], the compound BO2− is not thermodynamically stable. The anion B(OH)4− preferably forms. We propose thus a modified scheme in Fig. 3: it involves the latter borate.
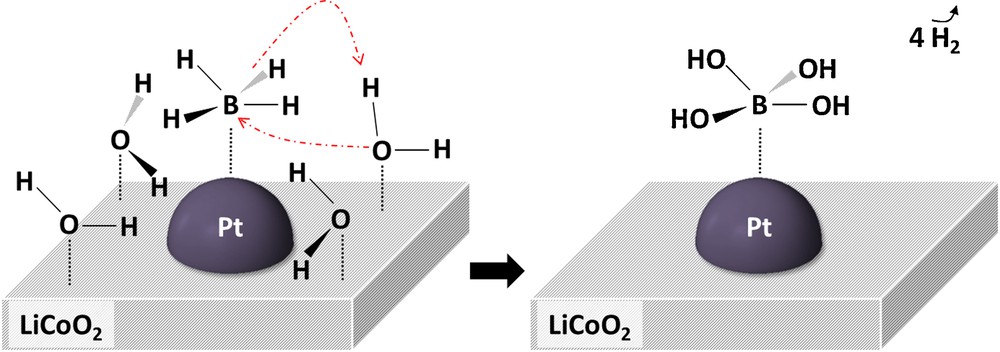
(Color online). Schematic representation of hydrolysis of BH4− and H2O on LiCoO2-supported Pt. This representation has been adapted from reference [48], but it has been modified in order to take into account the involvement of 4 H2O (instead of 2 H2O in the original figure) and the formation in hydrolysis.
Liu et al. [50] investigated the Pt–LiCO2–assisted hydrolysis of sodium borohydride also. They used X-ray absorption and confirmed the mechanism proposed by Kojima et al. For Co3O4-supported platinum, Hung et al. [51] reported a catalytic mechanism inspired from the previous one, where Co3O4 plays a role comparable to that of LiCoO2. The mechanism proposed by all of these authors is consistent with the Langmuir–Hinshelwood model where both reactants adsorb on the catalyst surface and react to give H2 and B(OH)4−.
Andrieux et al. [43] demonstrated the validity of the bimolecular Langmuir–Hinshelwood mechanism in their experimental conditions and proposed an appropriate reaction scheme for cobalt nanoparticle-catalyzed hydrolysis of sodium borohydride (Fig. 4). The catalytic process involves two different adsorption sites, denoted A and B. The sites A are suggested to be electron-rich elements such as Oδ− or Co0, whereas the sites B are electron-deficient, like Coδ+. The hydrolysis of the four hydrides of BH4− occurs one after the other by reaction with one adsorbed H2O for each hydride. However, the authors are questioning about the possible desorption of the reaction intermediates, the kinetics of the elementary steps, the nature of the sites A and B, and the nature of the catalytic surface. It is yet suggested that the hydrolysis of the four hydrides of BH4− would be realized with four different adsorbed H2O upon the adsorption of the anion. If we compare Kojima et al.’s mechanism [48] to Andrieux et al.’s [43], it stands out that the former group implicitly suggests that the A sites are metallic Pt and the B sites are electron-deficient Li or Co (Figs. 3 and 4).
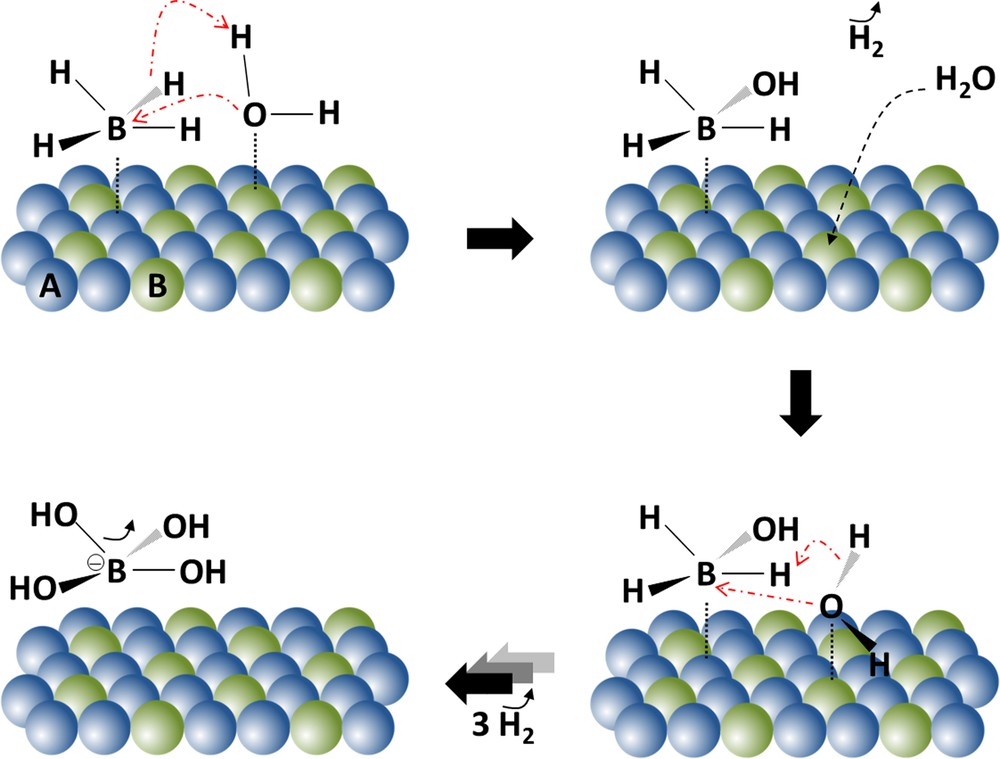
(Color online). Schematic representation of the Langmuir–Hinshelwood mechanism proposed by Andrieux et al. [43] to explain the surface activity of cobalt nanoparticles used as catalysts.
Adapted from [43].
3.2 Michaelis–Menten mechanism
In 2006, Guella et al. [47] reported an alternative mechanism. With the help of 11B NMR, it was proposed that the hydrolysis of BH4− is a stepwise reaction (Fig. 5). The first step is the dissociative adsorption of BH4− onto two Pd sites. It is supposed that one of the B−H bonds is homolytically broken during the dissociative adsorption; BH3− and H form. In the second step, adsorbed BH3− and adsorbed H react with one free H2O to form the intermediate BH3(OH)−, which desorbs, and H2. In the third and final step, BH3(OH)− undergoes hydrolysis with a faster rate than BH4− itself. The first and second steps of this mechanism are consistent with the Michaelis–Menten and Eley–Rideal models.
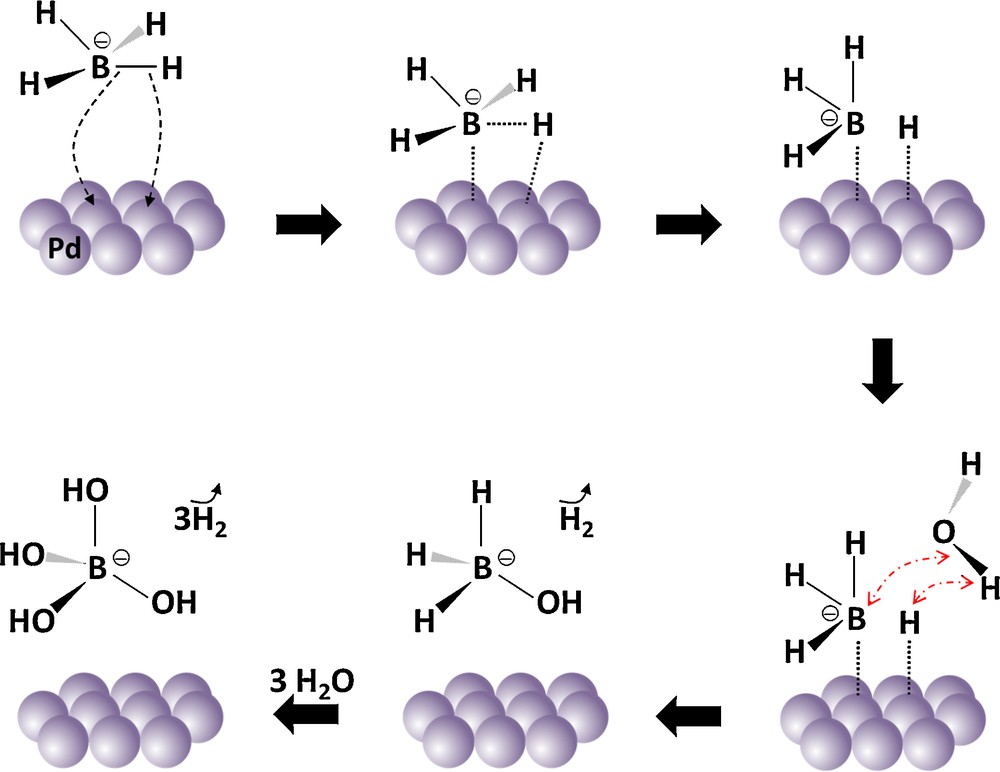
(Color online). Schematic representation of the Michaelis–Menten mechanism that could catch hydrolysis of BH4− over a Pd catalyst.
Adapted from reference [47].
A slightly different mechanism was proposed by Peña-Alonso et al. [52] for functionalized carbon nanotube-supported Pt or Pd (Fig. 6). It was proposed that BH4− undergoes a reversible dissociative adsorption onto two metal sites (MI and MII) leading to BH3− adsorbed on MI and H·adsorbed on MII. The negative charge of BH3− is transferred to the hydrogen thanks to the good conductivity of the carbonaceous support. The adsorbed hydride reacts then with one Hδ+ of one free H2O to liberate H2. The resulting OH− reacts with adsorbed BH3, while this BH3 transfers its remaining hydrides onto the free metal site MII. The adsorbed BH2(OH)− forms. The reaction continues according to a similar path until B(OH)4− and a total of 4 H2 form. A similar reaction sequence had been reported by Holbrook and Twist in the early 1970s for cobalt and nickel borides as catalysts and with heavy water D2O [53].
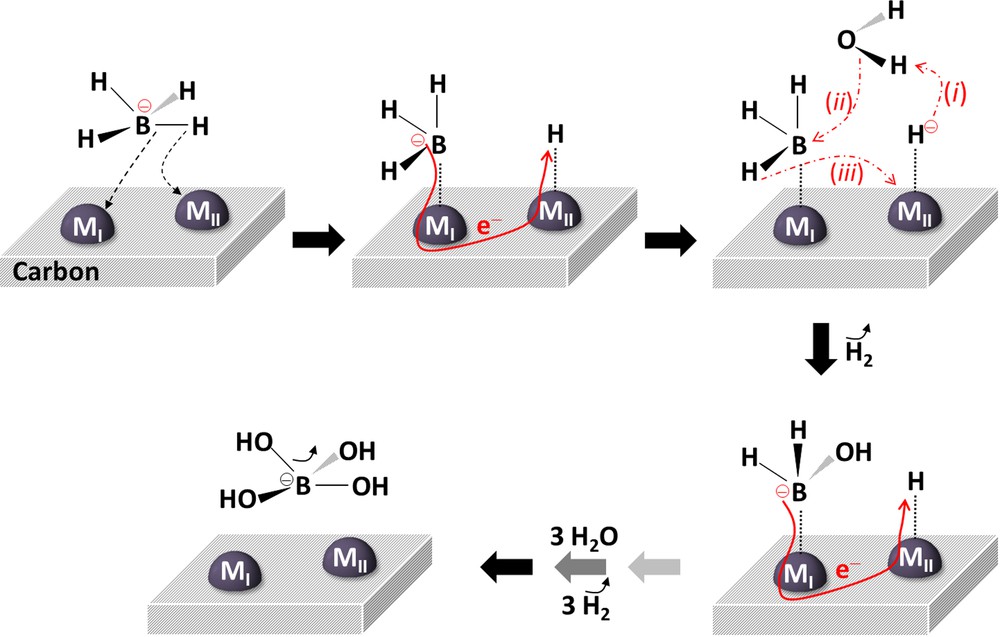
(Color online). Schematic representation of the Michaelis–Menten mechanism proposed by Peña-Alonso et al. [52] to explain the surface activity of supported Pt or Pd.
Adapted from [52].
Walter et al. [54] reinvestigated the aforementioned reaction sequence while proposing a rate expression taking into account a dependency versus the concentration of OH−. A reaction order of 1 was suggested and such an order was also proposed for the concentration of BH4−.
3.3 Summary
Despite numerous papers dedicated to hydrolysis of sodium borohydride (taken in liquid state as aqueous solution or in solid state), there are very few works proposing a detailed reaction mechanism. The most important ones have been reported above and only two mechanisms stand out, namely the Langmuir–Hinshelwood and the Michaelis–Menten (or Eley–Rideal, which also applies) ones. With the former model, it is believed that BH4− adsorbs on an electron-rich site like, e.g., Pt0, and that H2O adsorbs on an electron-deficient site like, e.g., cationic Co. With the latter model, only BH4− adsorbs on metallic sites and the adsorption is dissociative, with formation of BH3− and H·; the reaction of hydrolysis occurs with free H2O.
The above-mentioned works clearly show that identification of the catalyst surface sites is highly important in order to be able to propose detailed reaction mechanisms, especially when cobalt-based catalysts are used. For example, Andrieux et al. [43] suggest the presence of electron-rich and electron-deficient sites to support the Langmuir–Hinshelwood mechanism. However, it is important to mention that in all of these studies a reaction mechanism has been suggested, while the catalytic surface of the catalysts used had not been analyzed and determined.
4 Controversial cobalt-based catalysts
The kinetics of hydrolysis of aqueous solution of sodium borohydride depends on the nature of the metal catalyst and, especially, on its surface state. This is particularly the case when cobalt is used. In 2010, we showed that the nature of the catalytically active surface of cobalt is not unambiguously identified and that there is no study relevantly addressing the issue. We especially emphasized the difficulty to in situ (and operando) characterize the catalyst surface [15]. This is all the more arduous that the reaction medium is quite complex because of the presence of H2O, BH4−, Na+, OH−, possible reaction intermediates (though never evidenced), borate by-products B(OH)3/B(OH)4−, and polyborates [55]. Moreover, the catalyst surface evolves because of the strong reducing ability of sodium borohydride; the reduction generally takes place during the first seconds/minutes of the reaction, this period being defined as the lag or induction phase. A last problem is the possible adsorption and/or precipitation of borates on the catalyst surface [48].
In 2011, we demonstrated the formation of a core@shell-type structure, where the core is a cobalt compound (not yet clearly identified) and the shell (poly)borates (Fig. 1); such a structure is denoted Co@BαOβ(OH)γ [21]. For nickel-foam-supported cobalt, we showed that a (poly)borate layer strongly adsorbs on the surface via CoOB and BOB bonds and, together with the underlying cobalt compound, acts as a catalytic surface. However, this layer acts also as a poison when the catalyst is removed from the reaction medium. This layer acts also as a passivation layer. The initial catalytic activity can be recovered by washing the catalyst with an acidic solution in order to attack the (poly)borates layer and break the CoOB and BOB bonds. In doing so, the catalyst's surface finds itself in the state of the fresh material.
The role of cobalt in Co@BαOβ(OH)γ is not fully understood yet. Studies are in progress. However, it has been suggested [21] that the catalytic role of the surface layer BαOβ(OH)γ would be strongly related to the presence of underlying cobalt because of electronic effects. The H of the surface hydroxyl groups OH would be more protic than in, e.g., H2O or aqueous B(OH)4−; then the O of these hydroxyl groups would be electron richer. Hence, the first surface borates would be strongly bound to the metal surface. The electronic effect of cobalt would be nevertheless limited. After the formation of the BαOβ(OH)γ layer (whose thickness has still to be determined), the newly forming borates would not be surface adsorbed with enough strength and would desorb, rationalizing the observation of aqueous borates by solution-state 11B NMR [55].
Actually, our finding questions the various mechanisms reported to date, since none envisages the involvement of surface sites that would be different from metals or metal oxides. In our conditions, the catalytic surface would be the adsorbed (poly)borates in the form of a layer BαOβ(OH)γ. The interactions of the surface with the reactants could then take place via the electron-deficient B and/or H, and the electron-rich O. Besides, such surface species would be sensitive to the concentration of OH− and to the amount of water.
5 Hydrolysis on Co@BαOβ(OH)γ
It is worth noting here that the adsorption of H2O on the catalytic surface was evidenced while we were working on the methanolysis of sodium borohydride in the presence of supported cobalt catalysts [56]. It was shown that, for water–methanol mixtures, there is a competitive adsorption on the catalyst's surface. Water adsorbs more rapidly, but more strongly, which limits the reaction kinetics in comparison to that observed when pure methanol is used. Such better kinetics was also reported elsewhere [57].
On the surface of Co@BαOβ(OH)γ, the adsorption of the reactants cannot take place on cobalt sites, but on BαOβ(OH)γ. This layer of borates involves electron-deficient B, surrounded by likely 3 to 4 electron-rich O. With respect to the H involved in the surface hydroxyl groups, they are evidently protic. Hence, we believe that BH4− adsorbs on the surface layer through interactions between B of the reactant and O of surface OH. In all likelihood, the adsorption is dissociative, with formation of H2 and surface OBH3. This is the first step of the proposed hydrolysis mechanism (Fig. 7).
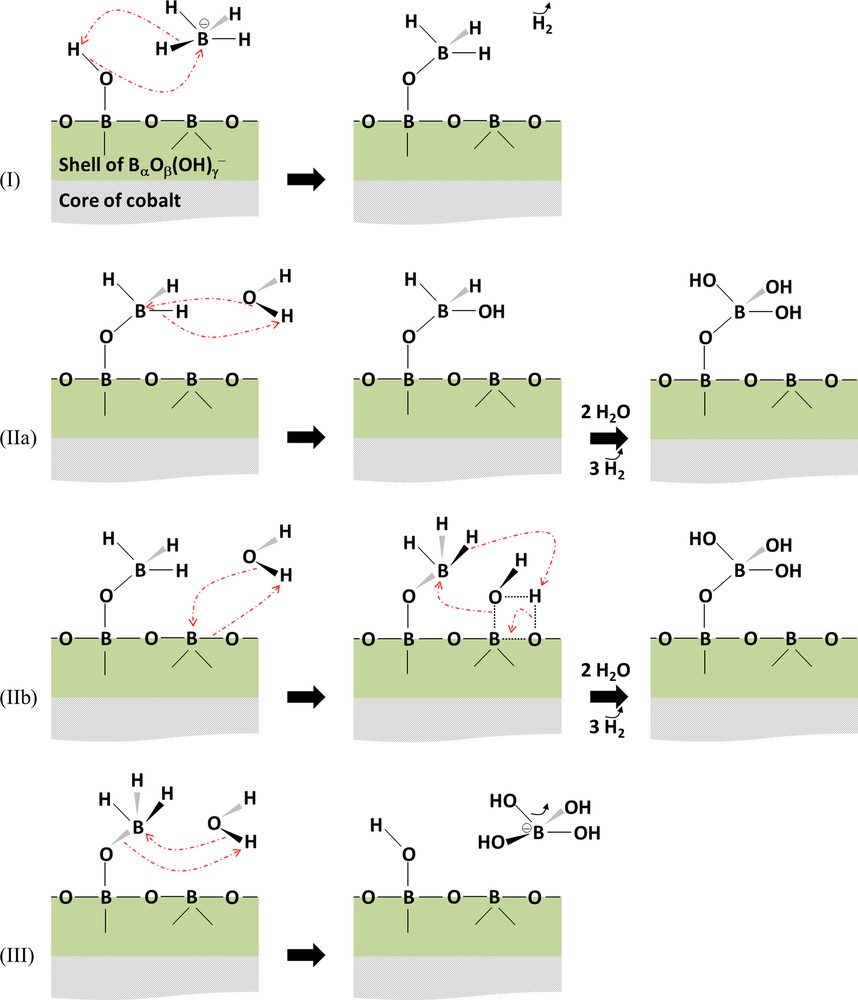
(Color online). Proposed mechanism for hydrolysis of BH4− on the surface of the layer BαOβ(OH)γ of the core@shell Co@BαOβ(OH)γ.
The second step is the hydrolysis of the remaining three hydrides of adsorbed BH3. There are in fact two possibilities (Fig. 7). The first one is the hydrolysis with free H2O, consistently with the Michaelis–Menten mechanism (Fig. 6) suggested by Peña-Alonso et al. [52]. The mechanism (Fig. 5) proposed by Guella et al. [47] is, in our opinion, unlikely, as we have never detected the formation of hydrolysis intermediates in our works dedicated to sodium borohydride [55] as well as to ammonia borane [58]. The second possibility is the hydrolysis of adsorbed BH3 with surface hydroxyl OH (whose formation would be favored by the presence of water in excess) or with adsorbed H2O. In this case, the reaction mechanism is consistent with the Langmuir–Hinshelwood model.
The third step is the desorption of adsorbed B(OH)3 by reaction with one free H2O. This leads to the liberation of the by-product B(OH)4−, making free the catalytic site through the formation of surface OH.
The mechanism in Fig. 7 should catch the different reaction orders (i.e. versus the concentration of BH4−, versus the amount of water and versus the amount of the catalyst) that have been reported to date. In addition, it should be able to predict the effect of the concentration of OH− as the catalytic surface Co@BαOβ(OH)γ should be dependent on the amount of OH− in solution. Liu et al. [39] reported slightly positive and then negative orders versus the concentration of OH− within the ranges 1–5 and 5–15 wt% NaOH, respectively. A negative order could be explained as follows. For the step 2a of the mechanism proposed in Fig. 7, high concentrations of OH− could be detrimental to the hydrolysis of adsorbed BH3 with free H2O because of a stabilization effect [7]. For the step 2b, the aqueous OH− could compete with H2O for adsorbing on the catalyst's surface. To justify the negative order they found, Liu et al. [39] conjectured that the anions OH− would compete with the transfer of BH4− to the catalyst surface. Further works are now required to construct a sophisticated kinetic model to fit the experimental data and predict the reaction mechanisms.
6 Conclusion
The reaction mechanisms of hydrolysis of aqueous alkaline solution of sodium borohydride are not fully understood yet and that is why, in the present review, we have surveyed and discussed the various mechanisms proposed to date. The mechanisms have been elaborated on the basis of kinetic studies using different models, i.e. the nth-order kinetics where n can be equal to 0, 1 or 2, the power law, the Langmuir–Hinshelwood model, the Michaelis–Menten model, and the semi-empirical kinetics. On the whole, the mechanisms proposed so far agree with the Langmuir–Hinshelwood or the Michaelis–Menten models. We have shown that the Eley–Rideal model could also explain the hydrolysis of sodium borohydride. Unfortunately, none of these kinetic models is able to completely describe the hydrolysis reaction. Discrepancies in the calculated kinetic parameters and contradictions in the mechanistic interpretations have arisen. For example, none of the models, and thus none of the reaction mechanisms proposed, catches the effect and the role of the hydroxide ions present in the aqueous solution of sodium borohydride. Such discrepancies and contradictions may be rationalized by the differences in the nature of the catalysts used, especially when cobalt-based catalysts are considered. It is indeed challenging to propose a reaction mechanism when the surface of the catalyst is not well identified. The current issue is therefore to unambiguously determine the catalytic surface involved in the reaction, i.e. how a sophisticated kinetic model could be proposed to fit the experimental data efficiently and predict the exact reaction mechanisms. Works are in progress in our group.