

1 Introduction
The direct conversion of methanol to methylal (or dimethoxymethane, or DMM) involves two successive steps:
- • the selective oxidation of methanol to formaldehyde (Eq. (1));
- • the condensation of the latter with two methanol molecules over an acidic function [1] (Eq. (2)):
| (1) |
| (2) |
Rhenium oxides, and particularly ReO3 in which the rhenium stands as Re6+, exhibits a catalytic activity favouring the direct conversion of methanol to methylal (DMM) [1]. Thus, the rhenium oxides retain both acidic and redox functions in the right balance for yielding DMM. The deposition of an oxorhenate layer over an oxide support of high surface area is a smart way to take advantage of the bifunctionality of rhenium oxide moieties together with increasing the rate and the cost-efficiency of the reaction. Many examples of supported oxorhenate catalysts devoted to various reactions can be found in the literature, among which ReOx/Al2O3 [2–7], ReOx/SiO2 [8,9] ReOx/TiO2 [2,10], ReOx/ZrO2 or Re/H-MFI [11]. Considering the catalytic properties of supported ReOx catalysts, the nature and structure of the support were found to exert a strong influence on the rate and orientation of the reaction [1,3,4]. In a previous work, we have proposed that the texture of the support as well as the hydration level would have a prime influence on the rhenium leaching and, hence, on the catalytic activity of supported ReOx materials [9,12].
The elucidation of the active phase of rhenium oxide species has attracted a significant amount of spectroscopic studies. In situ Raman spectroscopy is a technique of choice for investigating the structure of rhenium at molecular level. On the base of observation from Raman spectroscopy, it is generally agreed that supported rhenium oxides stand as isolated Re7+ moieties tetrahedrally coordinated with terminal and bridging oxo bonds [13–17]. Other techniques such as UV–visible absorption is relevant in the depiction of the oxidation state of rhenium as well as the detection of very small clusters retaining rhenium moieties at a reduced oxidation state [18–21]. It is to be noticed however, that to the best of our knowledge, no study has ever reported the UV spectrum of a titania-supported oxorhenate material. This matter of fact is certainly due to the strong absorption of TiO2 in the UV range, thus hiding all the bands coming from the rhenium oxide phase in this region. FTIR spectroscopy is of particularly relevance in investigating the OH groups [22] or the adsorption of probe molecules over the active centres of rhenium based materials. Other techniques such as XPS or XAS are very useful in specifying the oxidation state of the rhenium entities [5,23,24].
Among the possible ways to prepare ReOx/TiO2 materials, the present work reports on the catalysts afforded by oxidative redispersion of rhenium oxide over the titania support. The general procedure and some observations of the genesis of the active phase were presented in details in a previous paper of our group [12]. Briefly, the preparation was inspired by the work of Okal and co-workers devoted to γ-alumina-supported rhenium catalysts [16,25,26] and is consistent with the heat treatment of a mixture of metal rhenium and TiO2-anatase support under oxygen flow to favour the oxidation of rhenium and its deposition over the surface.
In the present work, we propose a multi-technique investigation of two 6.9wt.% ReOx/TiO2 catalysts prepared by oxidative dispersion of rhenium onto two anatase supports differing from their specific area. As the rhenium loading is constant, both materials exhibit different rhenium surface coverage and different structures can be expected at molecular level. Thus it was possible to link the catalytic activity measured to the structure of the active phase rather than to the rhenium loading. To our knowledge, this work is the first report on supported oxorhenate catalysts which crosses together such a combination of different spectroscopic techniques: in situ DR–UV–visible, operando Raman spectroscopy, operando FTIR and XPS recorded under vacuum after a selected treatment in a dedicated catalytic chamber.
2 Experimental and methods
2.1 Catalyst preparation
The anatase materials, kindly provided by Sachtleben and labelled Homkat K03 (93 m2·g−1) and Homkat F01 (254 m2·g−1) [10], were vigorously rinsed in distilled water and heated under dry air up to 400 °C to remove any trace of alkaline ions in the structure. This point was checked by elemental analysis (ICP) and XPS. The as-prepared supports were mixed with the desired amount of rhenium powder (Puratronic, 99.999%, Alfa Aesar) and manually grinded together in an agate mortar. The resulting light-grey mixture was then calcined for 6 h under pure O2 flow (20 cm3·min−1) up to 400 °C (heating rate: 1 °C·min−1). After this step, the catalysts have turned to white and the measured Re content was 6.9 wt.% for both catalysts. Because of the difference in the surface area of the supports, the ReOx/TiO2-F01, further denoted as ReF for brevity, features a surface coverage of 1.0 atom of Re per nm2 while the ReOx/TiO2-K03, further denoted as ReK for brevity, features a surface coverage of 2.7 atoms of Re per nm2. Upon rhenium enrichment, the BET surface was found to be decreased in both cases (Table 1). Other characterization results such as TEM pictures can be found in a previous paper [10].
BET specific area values obtained from N2 adsorption experiments [27] over the anatase-TiO2 support after the above-mentioned rinsing and re-calcination procedure and the corresponding catalysts.
| Sample | BET surface (cm2·g−1) |
| K03 | 93 |
| ReK | 84 |
| F01 | 254 |
| ReF | 200 |
2.2 Operando Raman and FTIR spectroscopies and in situ DR–UV–visible
After a catalyst pre-treatment at 350 °C under pure He (or Ar) flow (20 mL·min−1), the methanol was introduced in the spectroscopic cell by means of a He (Ar) flow (typically 19–21 mL·min−1) bubbling in a saturator equipped with a condenser which temperature is set to 3.8 °C to yield a methanol partial pressure of 5 kPa. According to the procedure applied, 5 kPa of oxygen were introduced in the feed to reach a He/CH3OH/O2 or Ar/CH3OH/O2 mixture with a molar distribution of 90/5/5. The parameters were adjusted to reach a GHSV value of 26 000 mL·h−1·gcat−1. After having passed through the catalytic bed, the reaction products were analysed using μ-gas chromatograph (SRA) equipped with a Poraplot U and a 5A molecular sieve columns and TCD detectors. The in situ spectral analysis was allowed by using an environmental spectroscopic chamber developed by Harrick equipped with a specific dome for each technique used. For Raman, the dome was a planar one equipped with a quartz window [28]. For UV and IR spectroscopies, the dome was the one especially devoted to each technique and commercially available at Harrick Scientific.
Micro-Raman spectra were recorded in working conditions at different temperatures and under selected atmospheres using the 532 nm second harmonic line of a Nd:YAG laser (laser power on the sample: 5 mW). A 50× microscope objective was used to focus the excitation beam (13.6 μm spot) and collect the scattered light. The scattered light was collected through a confocal hole (150 μm) by a nitrogen cooled CCD (Labram Infinity, JobinYvon). DRIFT spectra of the catalyst surface were recorded with 4 cm−1 resolution using a Thermo 460 Protege FTIR spectrometer equipped with a MCT detector.
UV/vis diffuse reflectance (DR) spectra were recorded by a Cary 400 spectrometer (Varian) equipped with a diffuse reflectance accessory (Praying Mantis; Harrick) and the above-mentioned reaction chamber connected to a gas-dosing system. The samples were diluted with α-Al2O3 to reduce light absorption. A very careful attention was paid for recording a baseline containing the very same amount of TiO2 as the catalyst in order to experimentally remove the contribution of anatase from the UV absorption.
2.3 X-ray-induced photoelectron spectroscopy
XPS experiments were carried out under ultra-high vacuum (∼10−10 Torr) in a Vacuum Generators Escalab 220XL spectrometer equipped with a monochromatic aluminium source (1486.6 eV) for excitation. A power of 300 W was applied to provide an 8-mm-diameter X-ray beam. The analyser was operated with constant pass energy of 40 eV. Binding energies (BE) values were referenced to the one of the Ti2p level (considered at 459 eV in anatase). Spectral decomposition of the experimental 4f5/2 and 4f7/2 rhenium core level peaks was carried out using a mixed Gaussian/Lorentzian peak profile using the CasaXPS software. Because of spin-orbit splitting, the peak separation between 4f5/2 and 4f7/2 was fixed to 2.4 eV and the area of the Re 4f5/2 photopeak was constrained to be as high as ¾ of the one of Re 4f5/2. The identification of the oxidation states of rhenium was based on BE values of Re 4f levels in reference rhenium oxide compounds reviewed by Okal et al. [23]. Samples were measured after direct transfer under UHV from the catalytic chamber to the analysis chamber.
3 Results and discussion
3.1 Reducibility of the oxorhenate phase: in situ DR–UV and XPS
The dehydrated ReK catalyst was subjected to a partial pressure of 5 kPa of methanol in argon and in the strict absence of oxygen. The rhenium active site is thus expected to be reduced and the methanol oxidation cannot be sustained because of the lack of oxygen to regenerate the catalyst. However, this experiment is useful in understanding the state of the rhenium reducible sites prior to regeneration. Hence, one piece of the catalytic cycle can be assessed. The UV–visible spectra of ReK catalyst upon increasing the temperature under an Ar/CH3OH flow is presented in Fig. 1.
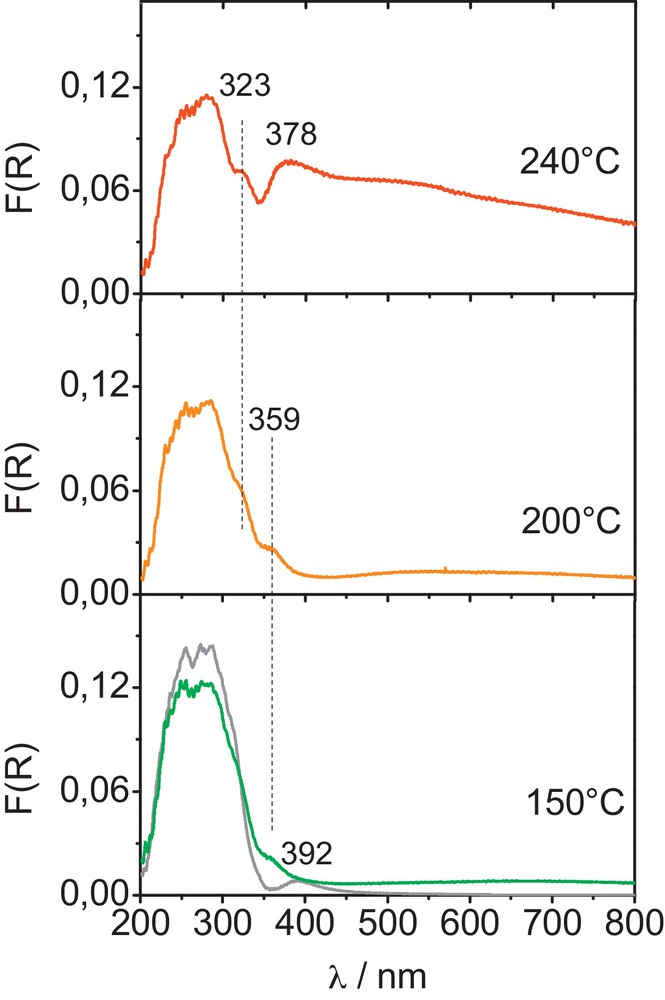
(Color online.) In situ UV–visible absorption spectra of ReK catalyst after exposure to Ar/CH3OH flow without oxygen at 150, 200 and 240 °C compared to the spectrum of dehydrated ReK recorded at 150 °C under pure O2 flow (grey line).
The absorption spectrum of the dehydrated ReK catalyst, shown in grey on Fig. 1, exhibits a broad absorption band in the UV region which can be described by three components around 220, 240 and 290 nm (Fig. 1, bottom). In addition, a small, yet significant band is also detected at 392 nm together with a very broad yet minor absorption centred around 550 nm. The absorption in the UV region is typical of distorted tetrahedral ReO4− moiety embedding Re7+ species while the one in the visible range essentially comes from reduced moieties [8,18–20].
Upon a reducing treatment under 5 kPa of methanol in argon at 150 °C, the absorption in the UV range is reduced while the absorption increases at 359 nm and, at a minor extend, in the visible range, increases. At 200 °C, the shoulder at 359 nm is still visible together with a new contribution at 323 nm. The increase of the temperature to 240 °C produced a drastic increase of the absorption around 400 nm with a remarkable contribution at 378 nm.
The assignment of the shoulders we have observed successively upon increasing the temperature under Ar/CH3OH flow is not straightforward. The bands observed between 320 and 400 nm are consistent with the DR–UV spectra collected by Edreva-Kardjieva and Andreev and assigned by the authors to a rhenium oxide-mesoperrhenate structure ReOx.Re7+O53− [8,18]. It was also shown by Stoyanova et al. that the relative contribution of this structure increases as the rhenium content increases [19].
The very broad absorption around 550 nm in the activated catalyst and after reduction under methanol up to 200 °C can be assigned to Re6+ while reduced species having an oxidation state comprised between 0 and +IV are responsible of the absorption around 400 nm and in the whole visible range [19].
The ReF catalyst features a level of surface coverage three times lower than the ReK one. The DR–UV spectrum of the ReF materials after calcination in O2 presented in Fig. 2a shows quite well-defined absorption bands at 216, 235, 260 together with shoulders detected at 286, 310 and, at a minor extent, around 400 nm. Contrary to what was observed in ReK material, no absorbance is visible around 500 nm, indicating that rhenium entities are mostly consistent with Re7+. The presence of sharp absorption bands related to Re7+O4 entities in distorted tetrahedral geometry observed at 216, 235 and 260 cm−1 is an indication of site isolation, which correlates the lowering of the rhenium surface coverage in comparison with ReK catalyst. Under 5 kPa of methanol in argon in the absence of oxygen, the trends are similar to those observed for ReK. However, the absorption in the UV range was found to be more pronounced at 150 °C in ReF in comparison with ReK, indicating that the reduction of rhenium VII+ was found to be more effective. At 200 °C, a significant amount of isolated Re7+ moieties are reduced to Re6+ (Fig. 2c) while more heavily reduced rhenium entities (Ren ≤ 4+) are detected after reduction at 240 °C under methanol (Fig. 2d). When allowing O2 in the feed and, thus, allowing the catalyst to be subjected to the genuine operating conditions, the spectrum of the oxidized phase is rapidly recovered and the absorption in the UV domain reaches 2/3 of the one registered before reaction (Fig. 2e).
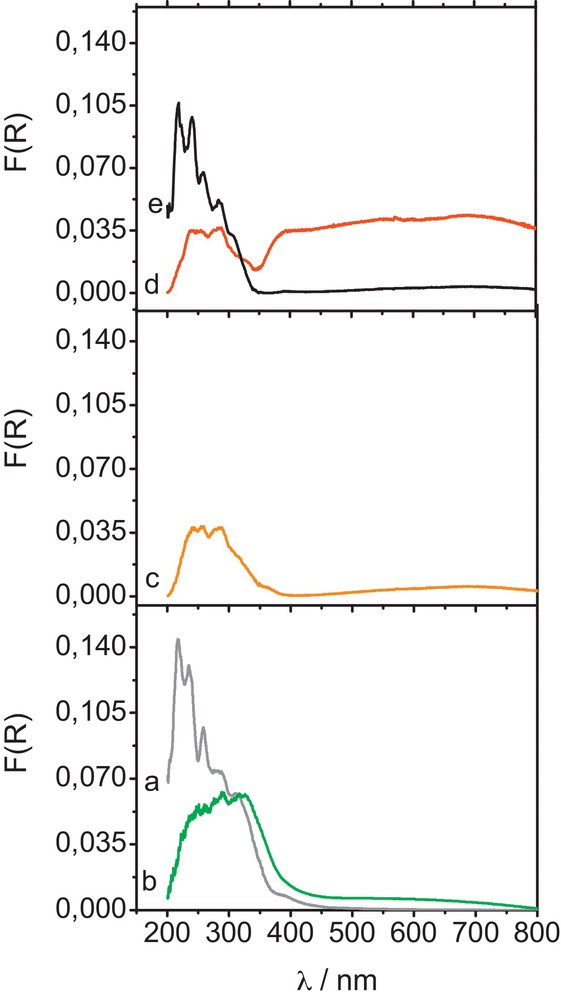
(Color online.) In situ UV–visible absorption spectra of ReF catalyst (a) under dehydrated conditions at 150 °C, recorded under pure O2 flow (b) after 1 hour reduction at 150 °C under Ar/CH3OH flow, (c) after 1 hour reduction at 200 °C under Ar/CH3OH flow, (d) after 1 hour reduction at 200 °C under Ar/CH3OH flow and (e) under operating conditions at 240 °C under Ar/O2/CH3OH flow.
Despite our efforts in adjusting the dilution for the recording of background, the strong absorption of the anatase support in the 200–400 nm range, however, makes it difficult to analyse precisely the structure of the oxorhenate phase at fully oxidised state. Moreover, the width of the absorption bands in the visible range under reducing atmosphere prevents any definite conclusion on the oxidation state of rhenium under methanol flux. In order to complete the information on the oxidation state of rhenium, photoelectron spectroscopy was applied after reductive treatment and reaction conditions without contacting the catalyst with air before analysis and the resulting XPS profiles of Re 4f5/2 and Re 4f7/2 core levels are presented in Fig. 3.

XPS spectra of ReF (a–c) and ReK (d–f) catalysts after subsequent treatments; experimental curves are the light-grey lines: a) and d): 1 hour at 240 °C under pure He flow; b) and e): 1 hour at 240 °C under He/CH3OH flow; c) and f): 1 hour at 240 °C under He/O2/CH3OH flow.
After activation in pure He, both catalysts retain a mixture of Re6+ and Re7+ with an approximate ratio of 10%/90% in the case of ReF and 20%/80% in the case of ReK (Fig. 3a and d). It is to be recalled, however, that the population of Re6+ might be overestimated due to the combined reducing effects of the ultra-high vacuum and the X-ray beam. Interestingly, the ReK catalysts seems to retain relatively more Re6+ moieties, as already suggested by DR–UV–visible spectroscopy. The spectral decomposition of the core level peaks recorded after one hour exposure to a He/CH3OH flow at 240 °C clearly indicates that a majority of rhenium sites features Re4+ sites. Indeed, the ReK catalyst embeds 53% of Re4+, 26% of Re6+ and 21% of Re7+. On the other hand, the ReF catalyst embeds 65% of Re4+, 35% of Re6+ and no more Re7+. It can be concluded from this result that ReF is more reducible than ReK, which is in good agreement with the conclusions of our DR–UV study and the work of Stoyanova et al. [19]. Under operating conditions (Fig. 3c and f), the rhenium is partly re-oxidised to a mixture of Re6+ (14% in ReF and 22% in ReK) and Re7+ (86% in ReF and 78% in ReK).
From the UV–visible and the XPS experiments, the isolated ReO4 entities which characterize the ReF catalyst are much more reducible under a flux of methanol carried in inert atmosphere.
3.2 Operando investigations along a reducing/operating procedure mimicking the conversion of methanol
We have investigated the evolution of the structure of the active phase under operating conditions using Raman spectroscopy and we have identified the adsorbed species using FTIR. At the same time, the methanol conversion as well as the identification and quantification of the products were measured by online GC analysis. The general procedure we have followed for both FTIR and Raman operando studies is summarised in Scheme 1.

Overview of the reducing/operating procedure applied for the operando investigations: a: 1-h stabilisation in He, 240 °C; b: 1-h reduction in He/CH3OH 200 °C; c: 1-h reduction in He/CH3OH 240 °C; d: 30-min rinsing in He, 240 °C; e: 1-h reaction under He/O2/CH3OH 240 °C; f: 30-min rinsing in He, 240 °C; g: 30-min-to-1-h rinsing in O2, 240 °C.
3.2.1 Methanol conversion during the operando Raman and IR investigations
We have followed the operando procedure described above using the same environmental chamber and the same online GC analysis for both the IR and Raman techniques. As a result, it was not surprising that the values featuring the products of methanol conversion were very similar for both experiments. The conversion levels as well as the selectivity are presented in Table 2. In order to allow comparison, results obtained at 240 °C from a catalytic test without reducing step in a plug-flow reactor and with the same velocity (GHSV = 26 000 mL·h−1·gcat−1) are also presented in brackets.
Conversion and selectivity levels measured along the reducing/operating procedure detailed in Scheme 1.
| Catalyst | Feed | Conversion (%mol) | Selectivity (%mol) | |||||
| DMM | DME | MF | F | CH4 | COx | |||
| ReF | He/CH3OH 200 °C | 2 | 0 | 7 | 66 | 0 | 0 | 27 |
| He/CH3OH 240 °C | 8 | 0 | 9 | 51 | 0 | 12 | 28 | |
| He/O2/CH3OH 240 °C | 10 (19) | 40 (71) | 5 (6) | 24 (22) | 31 (0.5) | 0 (0.5) | ||
| ReK | He/CH3OH 200 °C | 7 | 0 | 16 | 65 | 0 | 0 | 19 |
| He/CH3OH 240 °C | 12 | 0 | 16 | 25 | 0 | 17 | 42 | |
| He/O2/CH3OH 240 °C | 32 (28) | 75 (79) | 15 (14) | 8 (6) | 1,5 (0,5) | 0 (0) | 0,5 (0,5) |
Concerning the results of the catalytic test in a plug-flow reactor, the activity of ReK is higher than the one reported for ReF. Moreover, the ReF catalyst shows a higher level of oxidation and the formation of an increased share of methyl formate at the expense of the targeted DMM.
As far as the operando testing is concerned, at 200 °C under He/CH3OH, the main product yielded over both ReF and ReK catalysts is the methyl formate. The latter partially decomposed to CH4, and CO2 when the temperature rose up to 240 °C. The selectivity to DME remained relatively constant over increasing the temperature and close to 8 and 16% for ReF and ReK, respectively.
The conversion levels are expected to be much higher in the presence of oxygen. This was the case for ReK catalyst, whereas the conversion recorded over ReF catalyst remained low (10%) in comparison with what was measured (19%) during the catalytic test performed using a plug-flow reactor and without reducing step. Under He/O2/CH3OH mixture, DMM was found to be the main reaction product over both catalysts. In the case of ReF, still, the selectivity towards formaldehyde (31%) is significantly increased at the expense of the DMM production. This trend could be an indication of the poisoning of acidic functions of ReF during the reducing step. Indeed, this was not observed in the catalytic test without reducing treatment. In comparison with ReK, the ReF catalyst exhibits a stronger oxidative behaviour, as indicated by the formation of MF at the expense of DMM. This correlates the enhanced reducibility of the oxorhenate species in ReF in comparison with ReK.
3.2.2 Structure of the oxorhenate phase upon reduction under a partial pressure of 5 kPa of CH3OH and under operating conditions: operando Raman spectroscopy
The in situ Raman spectra of ReF and ReK catalysts along with the catalyst activation in pure helium at 350 °C followed a reducing step under 5 kPa of methanol in helium at 150, 200 and 240 °C and under operating conditions are presented in Fig. 4.

Evolution of Raman spectra of ReF (left) and ReK (right) catalysts during the operando procedure: a: calcined catalysts, ambient conditions; b: dehydrated material-spectrum collected under He flow at 150 °C, under He/CH3OH flow at (c) 150 °C, (d) 200 °C, (e) 240 °C; under He/O2/CH3OH flow at 240 °C for (f) 30 minutes and (g) 60 minutes; under pure O2 flow at (h) 240 °C and (i) back to room temperature. The “A” letters indicate the main structural bands of anatase. In insert (green colour online): magnification of the 1200–1700 region of ReF (f). Masquer
Evolution of Raman spectra of ReF (left) and ReK (right) catalysts during the operando procedure: a: calcined catalysts, ambient conditions; b: dehydrated material-spectrum collected under He flow at 150 °C, under He/CH3OH flow at (c) 150 °C, (d) 200 °C, (e) 240 °C; ... Lire la suite
The structure of the dehydrated catalysts as well as the evolution upon the successive treatment under methanol are essentially identical to the trends already reported by our group for similar catalysts prepared by impregnation of titania support by perrhenic acid [10]. In brief, the oxorhenate is featured by a peak around 1000 cm−1 and assigned to the symmetric stretch vibration of terminal ReO bonds. By comparison with the Raman spectrum of free perrhenate anion, it was proposed that this vibration is related to isolated ORe+7O3 entities having a more or less distorted tetrahedral geometry. Upon dehydration, this mode logically shifts toward higher wavenumbers in both catalysts. As for the impregnated materials we already reported on, unusual Raman scattering was detected around 600–900 cm−1 and 1300–1600 cm−1 which we have previously indirectly assigned to Re6+ species [10].
Under 5 kPa of methanol carried by helium, the Raman bands of both the anatase and the oxorhenate phase dramatically drop in intensity. In the case of ReF catalyst, the loss of Raman signal is particularly severe and two broad bands at 1330 and 1583 cm−1 of high intensity are respectively assigned to the D and G + D′ defect bands in carbon species [29] are observed, which are usually observed upon coke formation in spent catalysts [30]. On the other hand, the bands of anatase were still detected, together with a signal arising from the oxorhenate phase made of a sharp mode at ca. 480 cm−1 and a broader signal around 830 cm−1. Those modes are typical for the vibrations of ReO6 octahedra entering the structure of an orthorhombic crystal, as was observed by Kemmler-Sack et al. in the Sr2NaReO6 perovskite [31]. We have already proposed that the loss of Raman signal is typical of the ReO2 material retaining Re4+ species as stated by Kuzmin et al. [32]. This is in perfect agreement with the results from electronic spectroscopies. In addition, the presence of a disordered ReO2-like phase fairly correlates the modes detected at 480 and 830 cm−1 since ReO2 crystallises as ReO6 octahedra into an orthorhombic crystal structure.
Allowing oxygen in the feed and, hence, get to realistic operating conditions produced a partial recovering of the Raman signature of the catalyst. The coke is removed from the surface of ReF, but the signature of ORe+7O3 is still severely lower in the spectrum of the operating catalyst, as compared to the one recorded after activation. On the other hand, it is to be noticed that the broad features observed at 1300 and 1600 cm−1, which we have assigned to Re6+ are observed yet again in ReK. Those trends are in very good agreement with the results afforded from electronic spectroscopies.
3.2.3 Structure of surface species as probed by in situ FTIR under a partial pressure of 5 kPa CH3OH and under operating conditions
3.2.3.1 ReF catalyst
A procedure similar to what was applied for the Raman investigation was followed for giving insights on the adsorbed species under a reducing step under He/CH3OH followed by a so-called operating step under He/O2/CH3OH.
The DRIFT spectra recorded over the ReF catalyst are presented in Fig. 5. The activated material shows a sharp absorption band at 1617 cm−1 together with a broad band in the 2800–3500 cm−1 region and a sharper absorption at 3659 cm−1. The two first bands are respectively assigned to the bending and stretching modes of H2O chemisorbed onto the titania surface while the band observed at 3659 cm−1 (Fig. 6) arises from the OH stretch vibrations in TiOH groups located onto the (101) surface of anatase, which is the most stable one [33]. This observation indicates that in spite of the pre-treatment we performed at 350 °C and the fact that the oxorhenate appeared to Raman spectroscopy under a “dehydrated” form, physisorbed water is still present on the surface of anatase.
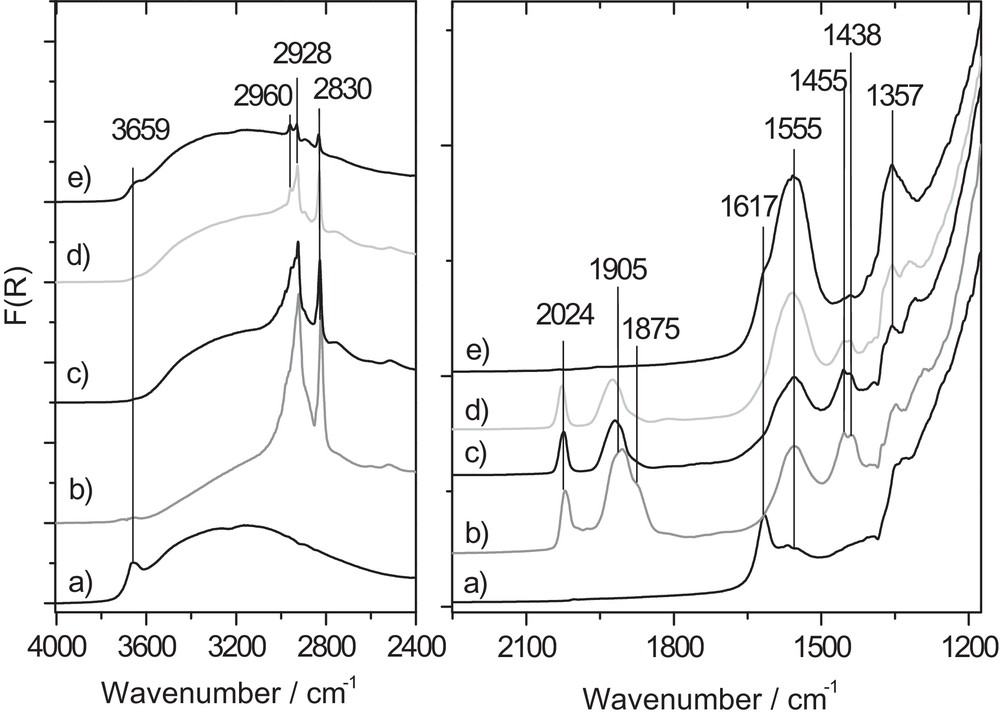
Evolution of DRIFT spectra of ReF catalyst at 240 °C under successive treatments: a): activated catalyst under He flow at 240 °C; b): 60 min under He/CH3OH flow (spectrum unchanged after 30′ rinsing in He at 240 °C); c): 60 min under He/O2/CH3 flow; d): after 30 min under He flow at 240 °C; e): regenerated 60 min under pure O2 flow at 240 °C.
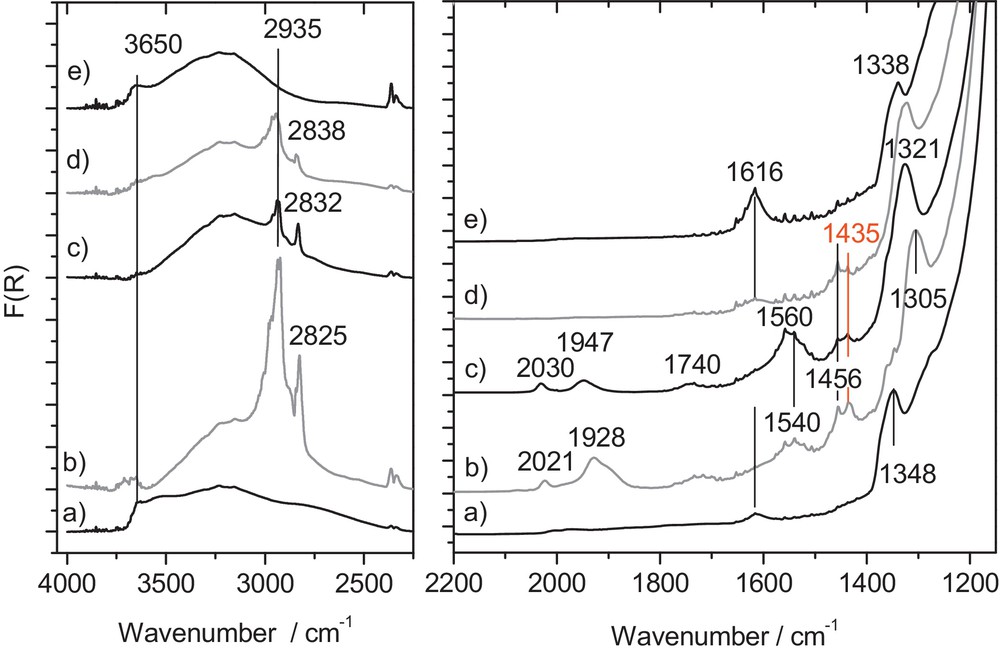
(Color online.) Evolution of DRIFT spectra of ReK catalyst under successive treatments: a): activated catalyst under He flow at 240 °C; b): 60 min under He/CH3OH flow at 240 °C; c): rinsed 30 min under pure He flow at 240 °C; d): 60 min under He/O2/CH3OH flow (spectrum unchanged after 30 min rinsing in He at 240 °C); e): regenerated 60 min under pure O2 flow at 240 °C.
Under He/CH3OH flux, new bands develop at 1438, 1455, 1555, 1875, 1905, 2024 cm−1 and in the 2800–2960 cm−1 CH stretch vibration region. Moreover, the OH stretch vibration region is modified, as the contributions of TiOH and H2O is visibly lowered. Allowing O2 in the feed to reach operating conditions induced a removal of the contribution observed at 1875 cm−1 observed in the absence of O2. Moreover, OH stretch modes assignable to physisorbed water are detected yet again in the 2800–3550 cm−1 region. After a 30-min purge at 240 °C under He, all the bands detected during the operating step are still present, indicating that the latter do not arise from the gas phase.
A subsequent treatment of the catalyst for 1 hour under 20 mL·min−1 of pure oxygen flow produced a removal of the bands between 1850 and 2025 cm−1 while the absorption at 1555 and 1357 cm−1 remained of high intensity. Moreover, residual bands of low intensities were still detected in the CH stretch vibration region (2800–2950 cm−1).
3.2.3.2 ReK catalyst
The same experiment was run over the ReK catalyst and the relevant in situ DRIFT spectra are shown in Fig. 6. The activated catalyst is characterised by a shoulder at 1349 cm−1, a band of low intensity at 1616 cm−1 and a very broad band between 2500 and 4000 cm−1.
After one hour at 240 °C under 5 kPa of methanol in helium, new bands at 2935, 2825, 2021, 1928, 1540, 1560, 1456, 1435 cm−1 are detected (Fig. 6b). Of those bands, only the doublet at 1540–1560 cm−1 exhibits an increase in intensity after rinsing in pure helium. However, all the features are still present at a lower extent after the purge in helium. The working catalyst under He/O2/CH3OH reveals a rather low concentration of adsorbed species with bands at 2838, 2935, 1616, 1456 and 1438 cm−1. Eventually, the surface was regenerated after a due flushing in O2 at 240 °C, still the absorption at 3500 and 1616 cm−1 appears stronger as compared to the one of the catalyst after activation. Eventually, a shoulder was detected at 1348 cm−1 and appeared to be reversibly shifted towards lower wave numbers under He/CH3OH flow (1305 cm−1) and He/O2/CH3OH/feed (1321 cm−1).
4 Discussion
4.1 Structure of the supported phase after activation in He and the evolutions induced by reducing and operating steps
The absorption of the ReF catalyst in the UV range is dominated by quite well-defined bands and the frequency of the terminal ReO bonds stretch vibration is rather high. Both techniques support the presence of ReO4 isolated species in which almost all rhenium entities feature an oxidation state +VII. On the other hand, it was shown by XPS, Raman and UV–visible absorption that ReK was found to retain a higher amount of small clusters embedding Re6+ moieties than ReF.
From both the in situ DR–UV and XPS measurements, it was evidenced that the isolated 4-fold coordinated species in distorted tetrahedral geometry are significantly more reducible under methanol than the small clusters. On the opposite, a fraction of the rhenium in ReK was found to be non-reducible, and we propose that this fraction is related to the small clusters above-mentioned. Indeed, for ReK catalyst, we have observed a gradual polymerisation of oxorhenate species under He/CH3OH, which was correlated to the presence of a non-reducible Re7+ population.
The reducing step prior to the reaction under normal operating conditions was found to exert a dramatic poisoning effect on the active phase in the low surface coverage catalyst (ReF) while the same reducing treatment was rather beneficial for the high surface coverage one (ReK). More precisely, the acidic function which catalyses the condensation of formaldehyde to dimethoxymethane is severely weakened. This trend definitely corroborates the essential difference in structure and reactivity of low and high surface coverage catalysts.
Unusual broad Raman bands were observed in the activated catalysts under inert atmosphere at ca. 650–900 cm−1 and 1200–1600 cm−1. Those Raman bands were found not to be stable under oxygen and to occur prior to methanol introduction. Upon excitation with another wavelength, the relative intensity of the modes at 600–900 cm−1 and 1300–1600 cm−1 is lower, but the observed wave numbers remain unchanged (not shown). As a consequence, fluorescence phenomenon was excluded, and we rather proposed that under dehydrated conditions and under inert atmosphere, both catalysts embed Re6+ involved in a disordered ReO3-like structure. Indeed, even though ReO3 exhibits no Raman-active mode, it was shown that upon disordering and passivation, two bands can be observed around 336 and 720 cm−1, possibly accompanied by a broad feature at 850–1000 cm−1 in polycrystalline powders and films [34]. Assuming the presence of a ReO3-like phase, we have previously proposed that the broad Raman bands observed at 1200–1600 cm−1 were assignable to second-order scattering, despite of the fact that the higher frequency modes are close to those of coke [10]. Indeed, ReO3 crystallizes in a cubic perovskite structure, which is well-known to favour the occurrence of second-order Raman scattering, such as in SrTiO3 [35]. Moreover, those second-order modes were already reported by Ishii and co-workers in ReO3 [36]. Eventually, our hypothesis is in good agreement with the presence of Re6+ in the activated catalyst stated using DR–UV–visible spectroscopy and XPS techniques. Similarly, even though the Raman signal of rhenium species is rather poor in ReF, a small contribution of this unusual scattering can be detected under He/CH3OH/O2 as well (Fig. 4f, in insert), correlating the presence of Re6+ suggested by XPS after reaction (Fig. 3c). It is to be noticed that this unusual scattering was already observed in catalysts afforded by the impregnation technique [10]. However, the relative intensities were significantly different, which prevents any quantitative analysis on the base of the Raman features. Eventually, the hypothesis we present here would require complementary and contradictory investigations.
4.2 Nature and evolution of the adsorbed species
The IR absorption bands we have observed during our experiments were thoroughly investigated and discussed in the literature, especially in studies devoted to the interaction of probe molecules with oxides.
A dissociative absorption of methanol onto an oxide surface leads to surface methoxy groups, which can be grafted either onto titania- or rhenium-containing groups. Busca et al. have investigated the adsorption of methanol over anatase surfaces by in situ FTIR and have concluded that methanol adsorbs dissociatively over TiO2 to produce methoxy species featured by absorption bands at 2965, 2930, 2830, 1452 and 1438 cm−1 [37,38]. The frequencies expected for rhenium methoxide are very close: 2955, 2930, 2849, 2821, 1456 and 1435 cm−1 [39]. After a careful comparison of the composition of bands in the region between 2800 and 3000 cm−1 and 1400–1450 cm−1, we rationally propose that the ReF under He/CH3OH flow mainly exhibits TiOCH3 species, while rhenium methoxy are the most abundant in ReK catalyst.
Formate groups HCO2− adsorbed onto the TiO2 (P25) surface were stated to be responsible of characteristic νAS(COO−) and νS(COO−) modes observed at 1560–1575 and 1360–1372 cm−1, respectively [38,40]. It is to be noticed that the ν(COO−) modes featuring formate moieties adsorbed on the supported rhenium oxide were also observed at 1550–1558 and 1356 cm−1 [39,41]. In the present study, a significant amount of formate is detected in the case of ReF and, at lower extent, of ReK catalyst. This is to be correlated with the production of methylformate as main reaction product observed by online GC.
Another set of bands are observed in ReF and, at a minor extent, ReK, around ∼1900–1930 and 2020 cm−1, which can be assigned to the ν(CO) stretch vibration in rhenium carbonyls [41,42]. It was proposed that the modes observed and 2020–2050 cm−1 are characteristic of carbonyls linearly bonded to rhenium species while the mode detected at 1860 cm−1 comes from CO vibration in the equatorial plane [42]. Those modes strongly support the presence of rhenium tricarbonyl moieties in a facial arrangement. The latter can be found in Re(CO)3(O–Alsupport)3 for example [43] and features a rhenium formal oxidation state of (+I). On the basis of our experiments, the formation of such species cannot be excluded. Indeed, rhenium tricarbonyl species are highly volatile and are expected to be removed from the sample upon pumping prior transfer to the XPS analysis chamber. Moreover, the formation of stable carbonyl species and the subsequent loss of rhenium by sublimation of Re+1(CO)3(L)3 – where L is an oxidic ligand – compounds upon oxygen introduction could characterize the deactivation of the low-loading material ReF. Indeed, the activity loss of ReF when a reducing step is applied prior to test under normal reaction conditions (with oxygen) is dramatic (in Table 2, values in brackets refer to those measured without reducing step). Concerning ReK, the formation of rhenium carbonyls is much less favoured while the intensity of the 2021 cm−1 band is severely decreased with regards to the one at 1928 cm−1, which could make sense to the formation of the rhenium tricarbonyl in its meridional isomeric form. In all cases, we already excluded the presence of remaining metallic rhenium in both dehydrated ReK and ReF catalysts in our previous work by means of DSC and TEM data [12].
Under operating conditions (in other words when flowing He/CH3OH/O2), both the rhenium carbonyl and formates remain on the surface of ReF from the DRIFT experiments. On the other hand, only TiO2-adsorbed formate species are still present on the ReK catalysts under operating conditions.
5 Conclusions
The two catalysts studied in the present work retain the same Re loading, yet differ from the rhenium surface coverage. At low surface coverage (ReF), the oxorhenate phase is consistent with a majority of isolated Re7+O4 species having a distorted Td coordination, as evidenced by UV–visible absorption. By increasing the surface coverage by a factor of three (ReK), clusters embedding Re7+ together with Re6+ were also present on the catalyst surface and those species were found to be poorly reducible under methanol. The presence of Re6+, favoured by the pre-treatment under inert atmosphere, is tentatively proposed to be responsible of the second-order Raman scattering observed between 1300 and 1600 cm−1. To the best of our knowledge, we bring here the first indirect observation of Re6+ using Raman spectroscopy in a working catalyst while the oxidation state of rhenium was certified using the appropriate technique under the very same conditions.
The increased reducibility of the rhenium at the surface of the low coverage catalyst has led to the formation of rhenium carbonyl under He/CH3OH flow, as shown by IR. Those species were removable under pure oxygen, but remained bonded to the rhenium phase under operating conditions. Moreover, the catalyst surface was poisoned by non-reacting carboneous species in the absence of oxygen, as evidenced by Raman spectroscopy. Stable formate species were also detected by FTIR during the reducing step, which were formed over rhenium and further deposited onto the acidic sites of titania.
From our operando investigations, we found that it is mandatory to rapidly re-oxidise the rhenium species, especially the isolated ones with O2 as co-reactant to avoid the formation of poisoning carbonyl and formate species leading to the formation of a coke layer. The regeneration of the catalyst is significantly favoured by the presence of small clusters retaining rhenium species at the +VI oxidation state and non-reducible Re7+ entities.
Acknowledgments
We acknowledge the French ministry for research and education for the PhD grant allocated to X. Sécordel and the IDECAT network funded by the European Commission under the Grant No. NMP3-CT-2005-011730.