

1 Introduction
Cancer is a very complicated group of diseases, which involves loss of cell growth control and cell alterations. One of the forms of cancer treatment consists of using chemical agents with cytotoxic properties. Research in this area is ongoing, particularly to synthesize anti-cancer agents which selectively target cancerous cells. The mode of action of one group of anti-cancer agents consists in blocking the template function of DNA. Control proteins mostly use the major groove while some polymerases and xenobiotics, among which antibiotics, bind to the minor groove. Examples of such minor groove binders include netropsin, distamycin, anthelvencyn, and kikumycin. Biochemical pharmacology research showed that the template functions of DNA are blocked by these oligopeptides with selective binding to AT sequences. Alteration of natural products to insure recognition of other DNA sequences have led to the production of compounds termed “lexitropsins” or information-reading molecules [1]. The binding of lexitropsins to the minor groove can occur via hydrogen bonds, since these compounds contain hydrogen-bond-accepting heterocycles, such as imidazole, furan or thiazole. In addition to hydrogen bonding, the binding of the lexitropsin to the minor groove can include electrostatic attractions. Van der Waals non-bonded contacts between the compound and the surface of the minor groove are carrying out the information-reading process.
Another mode of action of chemotherapeutic agents involves DNA alkylation. Among them are the mitomycins (Fig. 1) [2–4]. Their cytotoxic and antitumor activity is attributed to their ability to alkylate DNA monofunctionally and bifunctionally, the latter mode resulting in DNA interstrand and intrastrand crosslinks (ICL) [5]. Experimental evidence points to crosslinks being the lesions primarily responsible for the cytoxic properties of the mitomycins [6]. Their activity proceeds via a complex bioreductive process. On account of this property, MC is regarded as the prototype bioreductive antitumor agent [7]. However, a reductive inactivation pathway also prevails in tumor cells, diminishing the in vivo potency of MC and its alkylation and cross-linking of cellular DNA [8]. The partitioning of the active form of MC into reaction with DNA versus conversion to inactive metabolites depends on the kinetic competition of the two processes [9]. In contrast to most natural products, which interact with DNA, MC has no DNA-binding affinity until after its conversion to activated mitosene forms [10]. Therefore, it is likely that much of the MC that enters the cell is activated at intracellular locations, which are not in the vicinity of DNA. Flavoreductases that activate MC are present in the cytoplasm, in the microsomal fraction and in mitochondria, as well as the nucleus [11]. Consequently, in non-nuclear locations the active form is expected to decay mainly to inactive metabolites. In order to construct more potent anti-cancer agents, a rational approach may be to append mitomycin C with pre-activation DNA-binding capacity. Accordingly, we designed hybrid drugs where MC is linked to the N-methylpyrrole carboxamide framework present in lexitropsins. We hypothesized that on account of their affinity to DNA, the MC-pyrrole hybrids will be rapidly attracted to the cell nucleus, in the vicinity of DNA. This would make MC a more efficient DNA alkylating agent. The literature supports our hypothesis. A successful model for this scenario is the cross-linking of DNA by 2,3-bis(hydroxymethyl)pyrrole [12]. When this agent was conjugated to a distamycin derivative, the yield of DNA cross-linking increased 1000-fold [12]. The conjugate was also quite cytotoxic, in contrast to the marginally toxic parent compound. In addition, a monopyrrole conjugate of MC was found to be more efficient at cross-linking DNA in a pBR322 substrate [13].
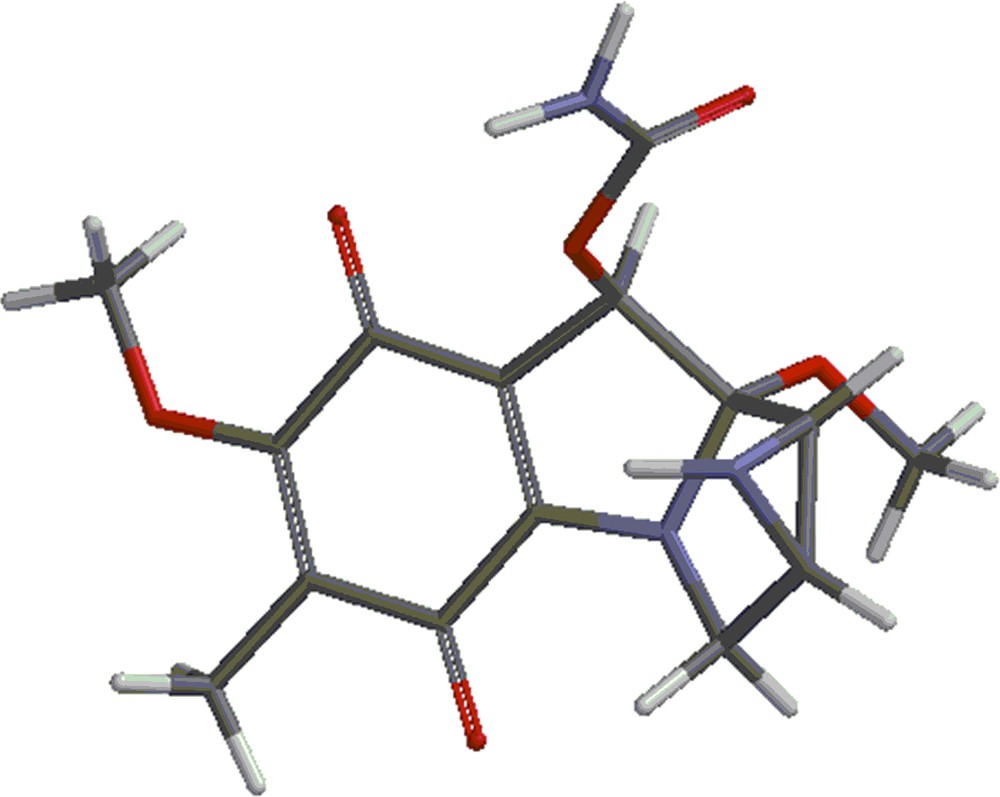
(Color online.) Mitomycin A.
Here, we describe the synthesis of MC-pyrrole carboxamide hybrids and theoretical calculations applied to the study of their synthesis.
2 Discussion
2.1 Theoretical calculations
Mitomycin A (shown in Fig. 1) was investigated with the DFT (density functional theory) using the 6-31G** basis set with the B3LYP method using the Spartan program [14].
The energies corresponding to the optimum geometries of the species shown in Figs. 1–3 were calculated. They are called respectively mitomycin A, structure 2 and structure 3. The optimum energies of the products of the reactions:
- • mitomycin a + structure 2 = structure 4 + methanol;
- • mitomycin a + structure 3 = structure 5 + methanol;
- • mitomycin a + structure 2 = structure 6;
- • mitomycin a + structure 3 = structure 7 were also calculated.
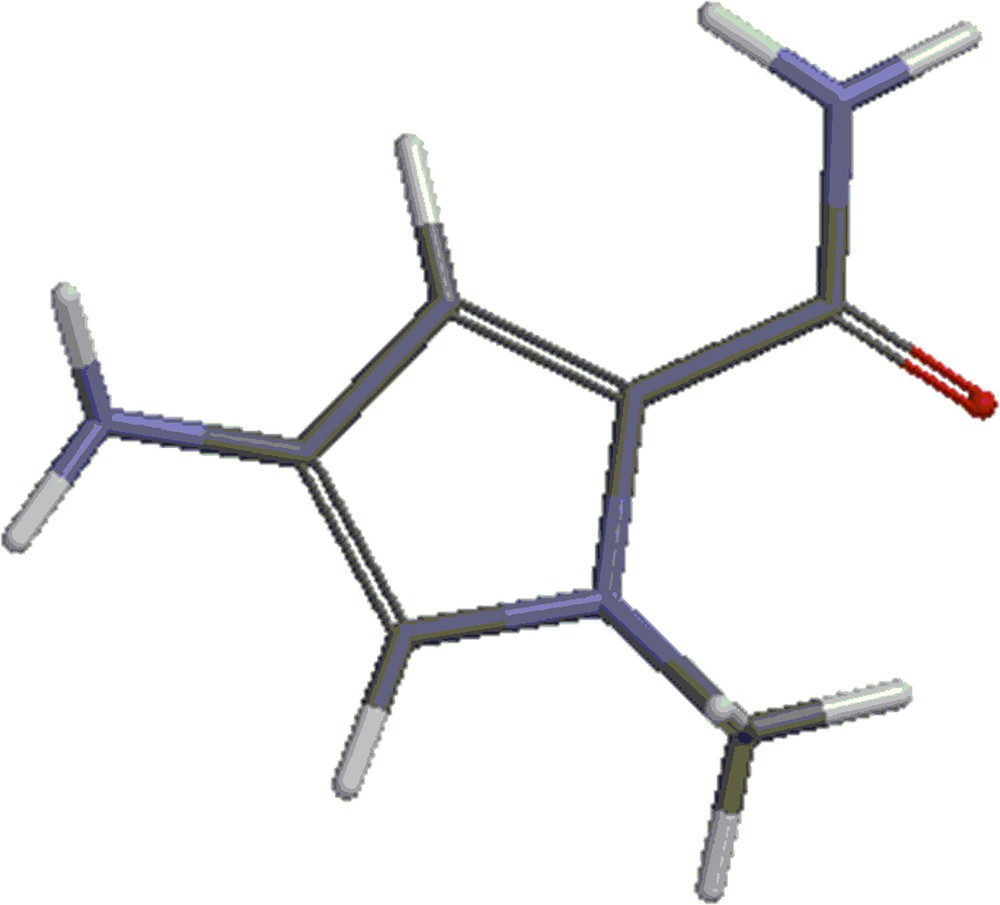
(Color online.) Structure 2: methyl-4-amino-1-methyl-1-H-pyrrole-2-carboxylate.
(Color online.) Structure 3: amine 2.
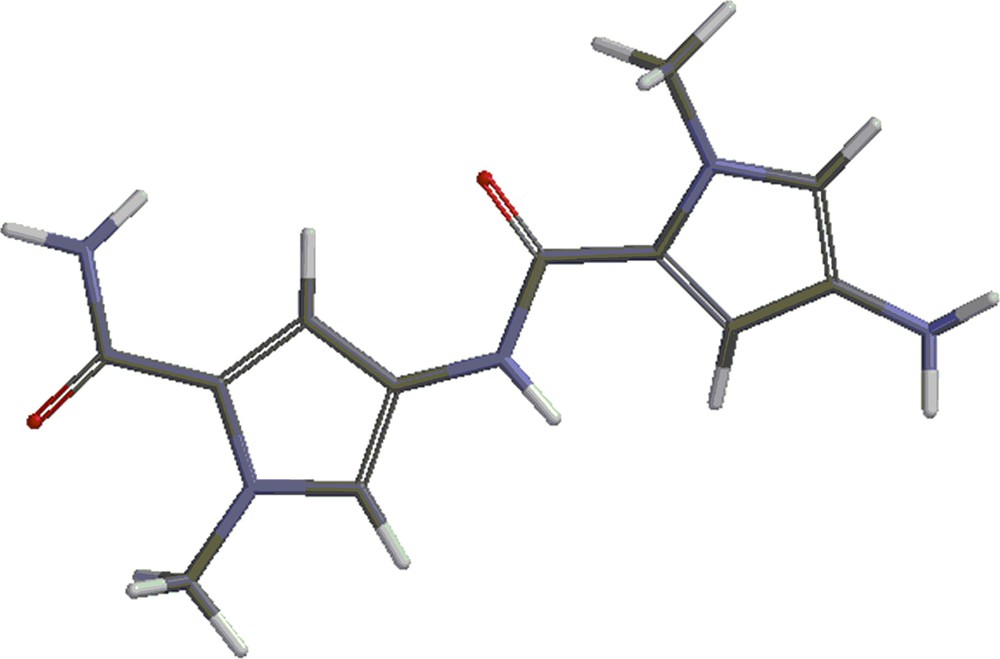
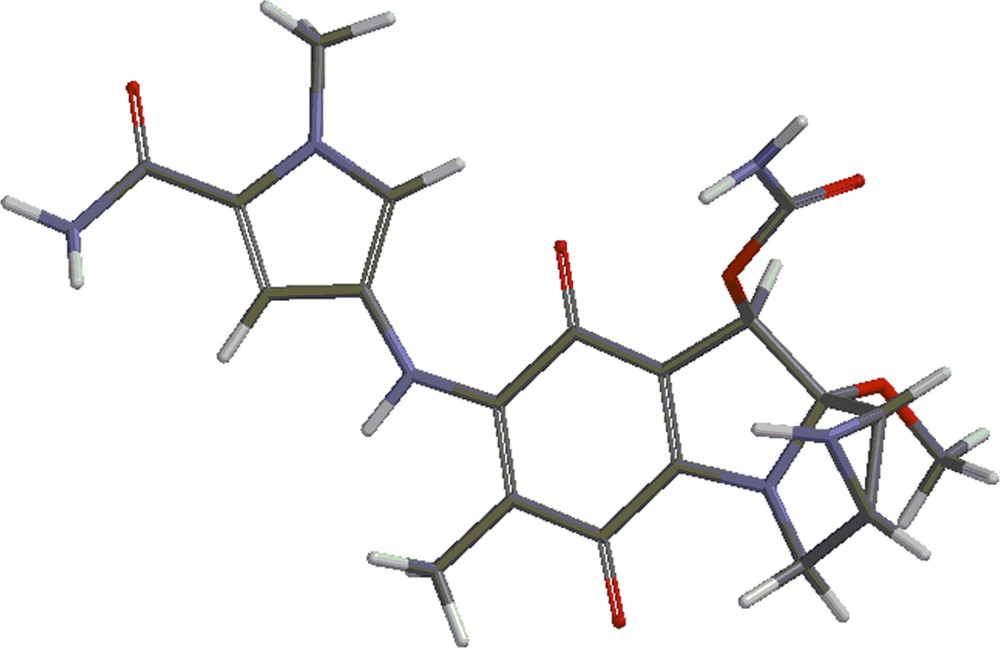
Structures 4 and 5 are shown in Figs. 4 and 5. Besides the covalent compounds resulting from the reactions 1 and 2, the calculations indicate the possible existence of complexes formed by mitomycin A and the lexitropsin fragments structure 2 and structure 3. These complexes are formed via hydrogen bonds and π–π interactions of aromatic nuclei. Figs. 6 and 7 show the complexes.
(Color online.) Structure 4.
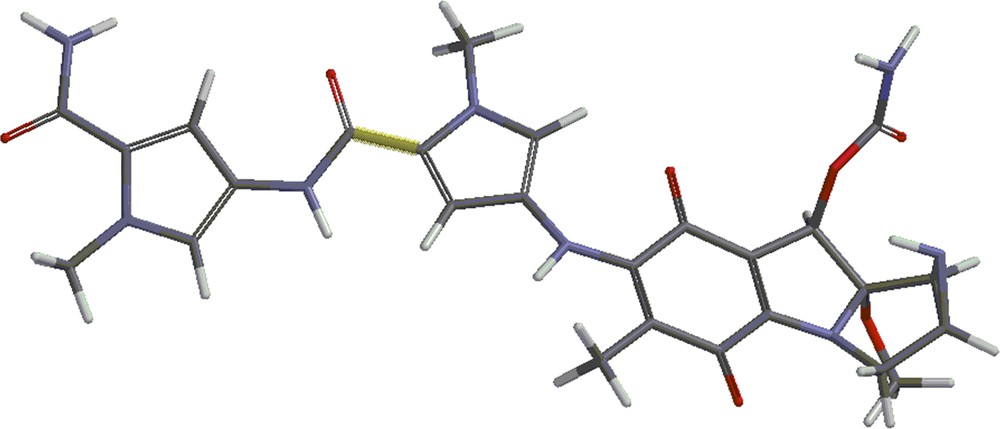
(Color online.) Structure 5.
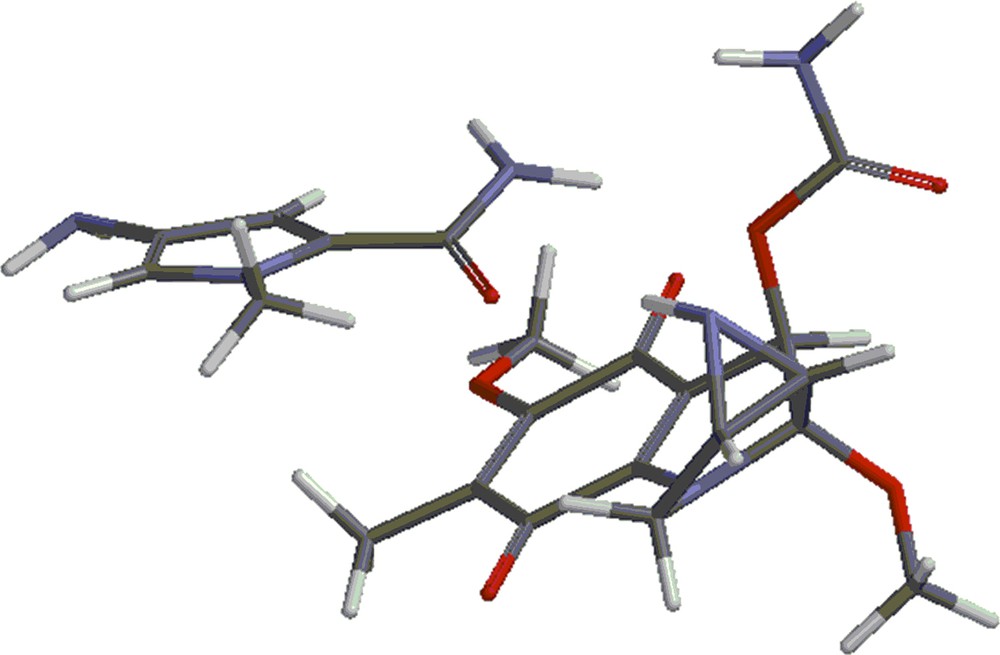
(Color online.) Complex formed by mitomycin A and structure 2.
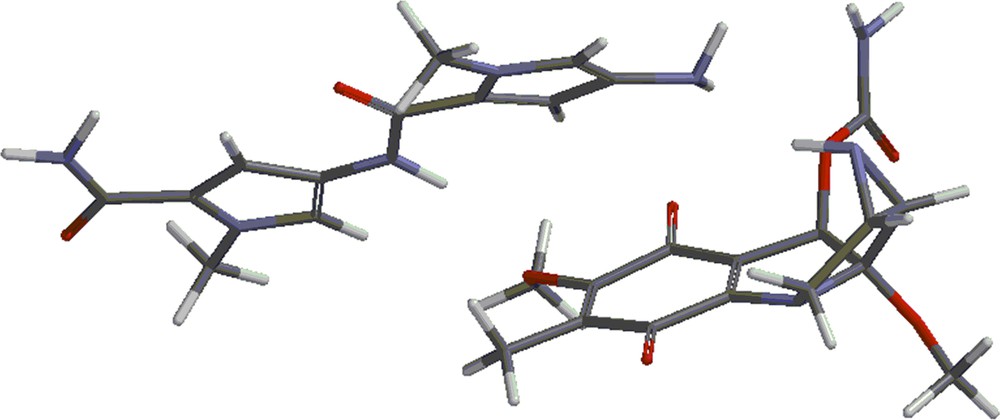
(Color online.) Complex formed by mitomycin A and structure 3.
Table 1 shows the energies of the different species in atomic units. Table 2 shows the energies of the above reactions. These energies were calculated by subtracting the energies of the reactants from the energies of the products and converting the result in kcal/mol.
Energies of the different species in atomic units.
| Compound | Energy (au) |
| Mitomycin A | –1197.38610 |
| Structure 2 | –473.55386 |
| Structure 3 | –890.55297 |
| Structure 4 | –1555.22041 |
| Structure 5 | –1972.21940 |
| CH3OH | –115.72400 |
| Structure 6 | –1670.95674 |
| Structure 7 | –2087.95213 |
Energies of reactions 1–4.
| Reaction | Energy (kcal/mol) |
| 1 | –2.46 |
| 2 | –3.01 |
| 3 | –10.50 |
| 4 | –8.16 |
As seen from Tables 1 and 2, the reactions of formation of the covalent compounds shown in structure 4 are exothermic, even though the energy obtained is small. These energies reflect the reactions taking place in the gas phase. As shown experimentally, in methanol as a solvent, the yield of the products is quite high, even though one of the products being methanol, an eventual equilibrium should be pushed toward the reactants. However, it is possible that methanol as a solvent gives rise to local interactions at different sites of the two reactants, interactions that would favour the reactions. Another difference between the gas-phase reactions and the reactions in solution is the fact that by addition of pyridine, the pH is slightly basic.
The complexes shown in Figs. 6 and 7 show stability, being lower in energy than the reactants by 10.50 kcal/mol and 8.16 kcal/mol, respectively. Methanol, the solvent in which the coupling reactions take place, might alter the formation of the complexes. Indeed, the complexes are formed partly via hydrogen bonds. These sites might be occupied by methanol molecules in solution and thus, prevent the formation of the complexes. That might explain the fact that the complexes were not detected as yet. They will form the subject of future studies.
2.2 Synthesis
The key step of linking the chemically fragile mitosane moiety to the N-methyl pyrroles was based on the preceding displacement of the 7-methoxy group of mitomycin A by aromatic primary amines, as described in Scheme 1 [15,16]. The MC-pyrrole hybrids (Figs. 8 and 9) were synthesized accordingly (methyl-4-amino-1-methyl-1H-pyrrole-2-carboxylate). HCl was treated with ammonia to yield free amine 1, the primary amine for the synthesis of the mono N-methyl pyrrole conjugate (Fig. 8). Amine 2, the primary amine for the synthesis of the bis-N-methyl pyrrole conjugate was obtained through DIC-mediated coupling of amine 1 and 4-(BOC-amino)-1-methylpyrrole-2 carboxylic acid followed by BOC deprotection via TFA treatment (Scheme 2). Incubation of each amine with mitomycin A in methanol for 16 h at room temperature yielded the desired MC-mono- (Fig. 8) and bis (Fig. 9)-N-methyl pyrrole conjugates in excellent yields, contrary to what DFT calculations predicted. All new structures were rigorously verified by HRMS, 1H and 13C NMR. The deep purple solution of mitomycin A in methanol (λmax = 218; 320, 524 nm) turned green after linkage of the mitosane moiety to the pyrrole fragment for both conjugates (λmax = 237; 382 nm for compound 1 and λmax = 238; 294, 372 nm for compound 2). This bathochromic shift is likely a consequence of the nature of the 7-subsituent: 7-aminomitosanes show a characteristic band at 360–380 nm, depending on the amine structure (360 nm for mitomycin C). On the other hand, 7-alkoxymitosanes absorb around 320 nm (316 nm for mitomycin A) [17]. Interestingly, two bands were observed in the UV–vis spectrum of the MC-mono N-methyl pyrrole conjugate, whereas the spectrum of MC-bis-N-methyl pyrrole conjugate exhibited three absorption bands.
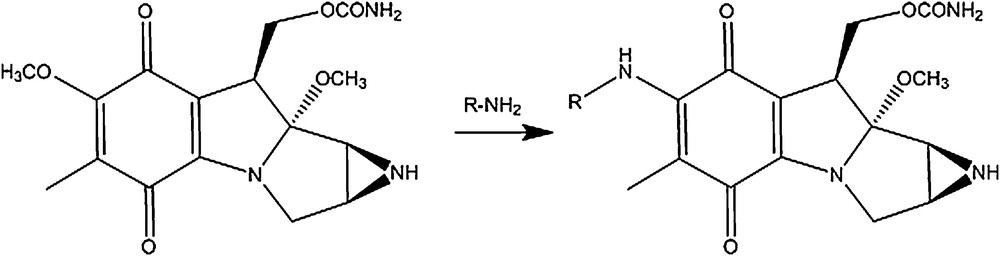
Displacement of the 7-methoxy group of mitomycin A by aromatic primary amines. R = aromatic group.
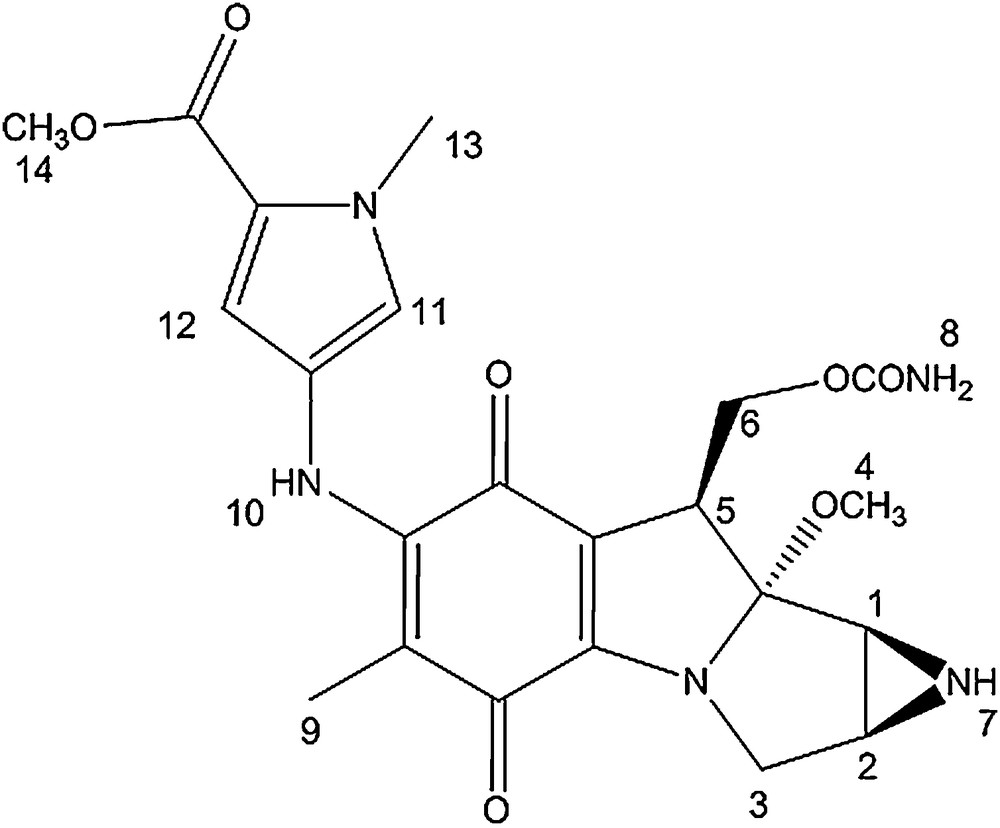
Mono N-methyl pyrrole conjugate.
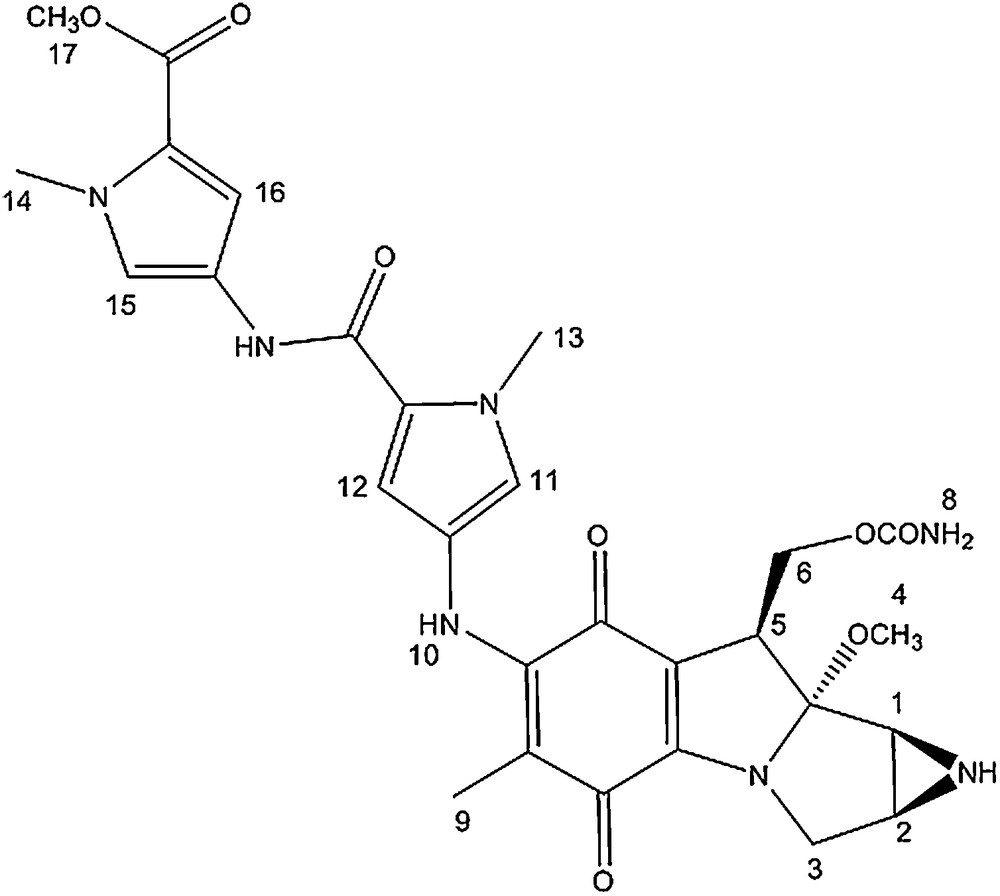
Bis-N-methyl pyrrole conjugate.
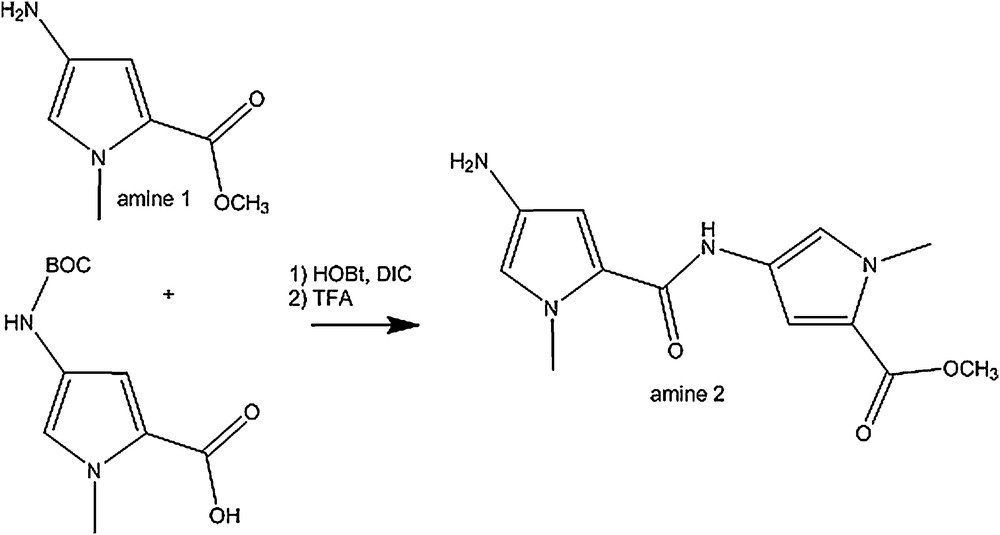
Synthesis of amine 2.
The 1H NMR spectra of the MC-mono and bis-N-methyl pyrrole conjugates are very similar. The signals corresponding to the mitomycin pyrrolo-indole core exhibit nearly identical chemical shits as that of mitomycin A, suggesting that appending pyrrole fragments at the C7 position has a meagre effect on the electronic environment of the mitosane protons. The pyrrole protons directly attached to the mitomycin fragment (11 and 12) showed a significant shift after the coupling reaction (δ = 6.70; 6.89 for compound 1 compared to δ = 6.42; 6.68 for the free amine and δ = 6.72; 6.83 for compound 2 compared to δ = 6.93 and 6.95 for the free amine).
In the 13C NMR spectra of both conjugates, signals from the pyrrolo-indole core appear at chemical shifts similar to that of mitomycin A. The main difference between the spectrum of mitomycin A and that of the conjugates is the signal corresponding to the quinone carbon directly attached to the methoxy group. It is shielded by around 10 ppm after coupling and linkage to the amino pyrrole group (δ = 143.9 and 145.6 for conjugate 1 and 2, respectively compared to δ = 154.4 for mitomycin A). In addition, the pyrrole carbons directly attached to the mitomycin fragment also showed a 1 to 4 ppm downfield shift after coupling.
3 Conclusion
We have constructed hybrid drugs where mitomycin C is linked to the N-methylpyrrole carboxamide framework present in lexitropsins. The coupling reactions leading to these products are efficient and yields are very high, although DFT calculations predicted the formation of other complexes. These complexes were not observed and could not be detected. A possible explanation for this fact is that the calculations were performed for the gas phase and that the reaction takes place in methanol instead. Consequently, the binding sites might be occupied by solvent molecules and prevent the formation of the complexes. An interesting spectroscopic feature of these hybrids is the red shift observed in the UV–vis spectrum. The next step is to test the properties of these derivatives against tumor cells.
4 Experimental
4.1 General remarks
Reagents were obtained from commercial sources and unless specified otherwise were used without further purification. Thin layer chromatographic analyses were carried out on 250-μm silica gel plates containing a fluorescent indicator. Column chromatographic purifications were performed using 200–300-mesh silica gel. 1H NMR spectra were obtained at 500 MHz at Columbia University and are referenced to residual protonated solvent. Chemical shifts are reported in parts per million and coupling constants are in hertz (Hz). UV–vis spectra were obtained on a Beckman Coulter DU spectrometer and HRMS were performed at the Mass Spectrometry and Proteomics Facility at the University of Notre Dame (Indiana, USA). HOBt: Hydroxybenzotriazole, DIC: N,N′-diisopropylcarbodiimide.
4.2 Synthesis of conjugate 1
Mitomycin A (9.00 mg, 0.026 mmol) was added to methyl-4-amino-1-methyl-1-H-pyrrole-2-carboxylate (3.97 mg, 0.026 mmol) in 0.2 mL of methanol and 0.04 mL of pyridine. The reaction was stirred under argon for 16 h at room temperature. TLC analysis revealed a new spot (rf = 0.3, SiO2, 5% methanol in dichloromethane). The new compound was isolated by preparative TLC (SiO2, 1% NH4OH, 4% methanol, 96% dichloromethane) and the green compound was dried in vacuo (10.8 mg, 88% overall yield).
4.2.1 1H NMR (methanol-d4)
δ = 1.52 (s, 3H, H9), 2.88 (d, 1H, J = 3 Hz, H2), 3.01 (d, 1H, J = 4.5 Hz, H1), 3.25 (s, 3H, H4); 3.50 (d, 1H, J = 12 Hz, H3), 3.64 (dd, 1H, J = 15, 4.5 Hz, H3), 3.81 (s, 3H, H14), 3.91 (s, 3H, H13), 4.17 (d, 1H, J = 13 Hz, H5), 4.34 (t, 1H, J = 10.5 Hz, H6), 4.73 (dd, 1H, J = 10.5, 4 Hz, H6), 6.70 (d, 1H, J = 2 Hz, H12), 6.89 (d, 1H, J = 2 Hz, H11).
4.2.2 13C NMR (methanol-d4)
δ = 8.3 (1C), 30.6 (1C), 34.6 (1C), 35.0 (1C), 47.2 (1C), 47. 9 (1C), 48.7 (1C), 55.1 (1C), 60.2 (1C), 104.4 (1C), 104.5 (1C), 108.1 (1C), 112.5 (1C), 118.9 (1C), 121.6 (1C), 123.7 (1C), 143.9 (1C), 153.8 (1C), 156.7 (1C), 159.9 (1C), 174.9 (1C), 178.2 (1C).
UV–vis: λmax = 237 nm; 382 nm.
HRMS m/z calcd C22H26N5O7 [M+H]+: 472.1832, found: 472.1827.
4.3 Synthesis of amine 2 (2)
Coupling reaction: HOBt (476 mg, 3.53 mmol) and DIC (482 μL, 3.08 mmol) were added to a solution of 4-(Boc-amino)-1-methylpyrrol-2-carboxylic acid (423 mg, 1.76 mmol) in 70 mL of dry dichloromethane. Methyl-4-amino-1-methyl-1-H-pyrrole-2-carboxylate (159 mg, 1.76 mmol in 1 mL of dry dichloromethane) was added slowly to the solution under argon. The reaction was left under stirring for 20 h. The solution was washed with water, dried (MgSO4), and the solvents were evaporated. The new compound was isolated by column chromatography (SiO2, 25% ethyl acetate, 75% dichloromethane) and the BOC protected amine 2 was dried in vacuo (497 mg, 75% overall yield).
4.3.1 1H NMR (methanol-d4)
δ = 3.81 (s, 3H), 3.84 (s, 3H), 3.91 (s, 3H), 6.73 (br, 1H), 6.90 (br, 1H), 6.93 (d, 1H, J = 2 Hz), 7.35 (d, 1H, J = 2 Hz).
4.3.2 13C NMR (methanol-d4)
δ = 28.7 (3C), 36.7 (1C), 36.9 (1C), 51.5 (1C), 105.8 (1C), 110.4 (1C), 119.6 (1C), 120.8 (1C), 122.4 (1C), 123.7 (1C), 123.8 (1C), 124.4 (1C), 158.9 (1C), 161.6 (1C) (1 peak missing, overlapping with the methanol peak).
HRMS m/z calcd C18H25N4O5 [M+OH]+: 377.1825, found: 377.1819.
Deprotection: the BOC protected amine 2 (100 mg, 0.27 mmol) was dissolved in 1.5 mL of dichloromethane and 0.5 mL of trifluoroacetic acid was added. The reaction was stirred at room temperature for 3 h. The mixture was diluted with dichloromethane and anhydrous sodium carbonate was added. The solution was filtered and solvents evaporated to give free amine 2 (66 mg, 90% overall yield).
4.3.3 1H NMR (methanol-d4)
δ = 1.53 (s, 3H), 3.81 (s, 3H), 3.91 (s, 3H), 3.96 (s, 3H), 6.93 (d, 1H, J = 2 Hz), 6.95 (d, 1H, J = 2 Hz), 7.07 (d, 1H, J = 2 Hz), 7.38 (d, 1H, J = 2 Hz).
4.3.4 13C NMR (methanol-d4)
δ = 35.5 (1C), 35.8 (1C), 50.1 (1C), 106.5 (1C), 108.9 (1C), 119.6 (1C), 121.1 (1C), 121.4 (1C), 122.1 (1C), 127.1 (1C), 155.7 (1C), 161.4 (1C), 163.0 (1C).
HRMS m/z calcd C13H17N4O3 [M+H]+: 277.1301, found: 277.1295.
4.4 Synthesis of conjugate 2
Mitomycin A (28.0 mg, 0.080 mmol) was added to amine 2 (22.0 mg, 0.026 mmol) in 0.5 mL of methanol and 0.1 mL of pyridine. The reaction was stirred under argon for 16 h at room temperature. TLC analysis revealed a new spot (rf = 0.2, SiO2, 5% methanol in dichloromethane). The new compound was isolated by preparative TLC (SiO2, 1% NH4OH, 5% methanol, 94% dichloromethane) and the green compound was dried in vacuo (40.0 mg, 84% overall yield).
4.4.1 1H NMR (methanol-d4)
δ = 1.57 (s, 3H, H9), 2.88 (d, 1H, J = 3 Hz, H2), 3.00 (d, 1H, J = 4.5 Hz, H1), 3.26 (s, 3H, H4); 3.53 (d, 1H, J = 13 Hz, H3), 3.64 (dd, 1H, J = 15, 4.5 Hz, H3), 3.81 (s, 3H, H17), 3.91 (s, 3H, H14/13), 3.93 (s, 3H, H14/13), 4.18 (d, 1H, J = 13 Hz, H5), 4.32 (t, 1H, J = 10.5 Hz, H6), 4.75 (dd, 1H, J = 10.5, 4 Hz, H6), 6.72 (d, 1H, J = 2 Hz, H16), 6.83 (d, 1H, J = 2 Hz, H12), 6.94 (d, 1H, J = 2 Hz, H15), 7.36 (d, 1H, J = 2 Hz, H11).
4.4.2 13C NMR (methanol-d4)
δ = 9.7 (1C), 32.3 (1C), 35.6 (1C), 35.7 (1C), 36.1 (1C), 43.2 (1C), 48. 7 (1C), 49.4 (1C), 50.1 (1C), 61.7 (1C), 105.9 (1C), 106.1 (1C), 108.9 (1C), 109.6 (1C), 109.9 (1C), 119.5 (1C), 121.1 (1C), 122.3 (1C), 122.7 (1C), 123.7 (1C), 123.8 (1C), 145.6 (1C), 155.4 (1C), 158.1 (1C), 159.6 (1C), 161.6 (1C), 176.4 (1C), 179.6 (1C).
UV–vis: λmax = 238 nm; 204 nm; 372 nm.
HRMS m/z calcd C28H32N7O8 [M+H]+: 594.2312, found: 594.2307.