

1 Introduction
Sulfur ylides R2S = C (R′) (R′′) (R, R′, R′′ = alkyl or aryl groups) are very reactive species with interesting applications in organic synthesis [1–4]. The juxtaposition of the keto group and carbanion in the α-keto stabilized sulfonium ylides permits a resonance delocalization of the ylidic electron density, providing additional stabilization to the ylide species. This allows the potential to act as an ambidentate ligand and thus to bond to a metal center through the carbanion (b) or the enolate oxygen (c). The enolate form (c) may assume either a cis or trans arrangement, the geometry of which will be retained upon bonding to the metal. Herein, we report the coordination of a sulfur ylide to a mercury(II) center through carbon and oxygen donor atoms simultaneously (Scheme 1).
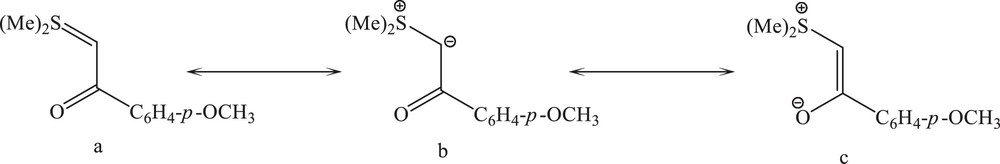
The canonical forms of (Me)2SCHC(O)C6H4-p-OCH3 (Y).
In contrast to C- and O-coordinated α-keto stabilized phosphorus ylides [5–8], there are few examples of transition metals coordinated to sulfonium analogs. On account of our knowledge to date, all previously reported sulfonium ylide complexes with soft metal ions e.g., Pd(II), Pt(II), Hg(II) and W(CO)5 are C-coordinated to the metal [9–11], and the only O-coordinated complex is [W(CO)4(C5H4NC(O)CHSMe2)] [12]. The coordination mode was not based on a crystal structure determination.
The synthesis of complexes derived from sulfonium ylides and mercury (II) halides was first reported in 1975 by Weleski et al. [13], who proposed a symmetric halide-bridged binuclear structure. In 1984, Tewari et al. [11] reported the synthesis of a series of transition metal halide complexes with various sulfonium ylides without further characterization. They reported complexes [{HgCl2(Y)}4]n (1) and [HgBr2(Y)]2 (2) as C-coordinate, presumably containing bridging X− groups, whereas herein we report on the pseudo five-coordinate Hg(II) polymeric structure for complex 1 and show an asymmetric halide-bridged binuclear structure for complex 2. Also, the characterization of 1H- and 13C NMR spectra of all complexes are presented, and their structures are compared with those of the analogous phosphorus ylides.
2 Experimental
2.1 Materials and physical measurements
All solvents were reagent grade and used without further purifications. NMR spectra were obtained with 400 MHz Varian MR400 and 90 MHz Jeol spectrometers in DMSO-d6 and CDCl3 as the solvent. Chemical shifts (δ) are reported relative to internal TMS (1H and 13C). Melting points were measured on a Stuart SMPI apparatus. Elemental analyses for C, H and N were performed using a PerkinElmer 2400 series analyzer. Fourier transform infrared spectra were recorded on a Shimadzu 435-U-04 spectrophotometer, and the samples were prepared as KBr pellets.
Data collection from suitable crystals of 1 was performed on an Oxford Diffraction single-crystal X-ray diffractometer using mirror monochromated Mo Kα radiation (0.71073 Å) at 130 K. Gaussian absorption correction was carried out using a multifaceted crystal model, using CrysAlisPro [14]. The analysis of the crystal structure of 2 was performed on a STOE IPDS-II diffractometer at 298 K, using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data collection was performed using the ω-scan technique and the STOE X-AREA software package [15]. The crystal structure was solved by direct methods and refined by full-matrix least-square method on F2 using the SHELXTL-97 [16,17] and X-STEP32 [18,19] crystallographic software package for 1 and 2, respectively. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at the calculated positions using a riding mode with fixed thermal parameters.
2.2 Synthesis of ylide (Me)2SCHC(O)C6H4-p-OCH3(Y)
2.2.1 (Me)2SCHC(O)C6H4-p-OCH3(Y)
To an acetone solution (10 ml) of dimethyl sulfide (0.062 g, 1.00 mmol) was added 2-bromo-4′-methoxy acetophenone (0.211 g, 1.00 mmol), and the mixture was stirred for 12 h. The solid product (sulfonium salt) was isolated by filtration, washed with ether and dried under reduced pressure. Further treatment with an aqueous 10% NaOH solution led to the elimination of HBr, giving the free ligand Y [20]. Yellow solid; yield: 76%, mp 130–132 °C. Anal. calcd. for C11H14O2S (%): C, 62.83; H, 6.71; S, 15.25. Found: C, 62.71; H, 6.79; S, 15.28. IR (KBr disk): ν (cm−1) (assign.): 1583(CO) and 857 (S+–C−). 1H NMR (CDCl3) δH ppm (multiplicity of the signal, integr., assign.): δH 2.96 (s, 6H, S(CH3)2), 3.79 (s, 3H, OCH3), 4.27 (1H, CH), 6.83 (d, 3JHH = 8.0 Hz, 2H, Ph), 7.74 (d, 3JHH = 8.0 Hz, 2H, Ph). 13C NMR (CDCl3, ppm) δC: 28.71 (s, S(CH3)2), 50.54 (s, OCH3), 55.23 (s, CH), 113.10 (s, Ph(p)), 127.95 (s, Ph(m)), 133.45 (s, Ph(o)), 160.90 (s, Ph(i)), 182.77 (s, CO).
2.3 Synthesis of the Hg(II) halide complexes
2.3.1 General procedure
To a methanolic solution (15 mL) of HgX2 (0.500 mmol) was added a methanolic solution (10 mL) of ylide Y (0.50 mmol). The mixture was stirred for 4 h. The separated solid was filtered and washed with diethyl ether.
2.3.1.1 Data for [{HgCl2(Y)}4]n (1)
White solid; yield: 98%, mp 205–206 °C. Anal. calcd. for Hg2Cl4O4S2C22H28 (%): C, 27.42; H, 2.93; S, 6.66. Found: C, 27.68; H, 3.01; S, 6.71. IR (KBr disk): ν(cm−1)(assign.): 1649 (CO) and 844 (S+–C−). 1H NMR (DMSO-d6) δH ppm (multiplicity of the signal, integr., assign.): 2.91 (s, 6H, S(CH3)2), 3.85 (s, 3H, OCH3), 5.42 (s, 1H, CH), 7.03 (d, 3JHH = 8.1 Hz, 2H, Ph), 7.76 (d, 3JHH = 8.1 Hz, 2H, Ph). 13C NMR (DMSO-d6, ppm) δC: 27.03 (s, S(CH3)2), 56.04 (s, OCH3), 65.29 (s, CH), 114.19 (s, Ph(p)), 127.67 (s, Ph(m)), 130.44 (s, Ph(o)), 163.68 (s, Ph(i)), 191.71 (s, CO).
2.3.1.2 Data for [HgBr2(Y)]2 (2)
White solid; yield: 91%, mp 200–202 °C. Anal. calcd. for Hg2Br4O4S2C22H28: C, 23.15; H, 2.47; S, 5.62. Found: C, 23.01; H, 2.29; S, 5.47. IR (KBr disk): ν(cm−1)(assign.): 1638 (CO) and 824 (S+–C−). 1H NMR (DMSO-d6) δH ppm (multiciplicity of the signal, integr., assign.): 2.89 (s, 6H, S(CH3)2), 3.82 (s, 3H, OCH3), 5.36 (s, 1H, CH), 6.88 (d, 3JHH = 8.1 Hz, 2H, Ph), 7.78 (d, 3JHH = 8.1 Hz, 2H, Ph). 13C NMR (DMSO-d6, ppm) δC: 27.03 (s, S(CH3)2), 56.13 (s, OCH3), 65.46 (s, CH), 114.23 (s, Ph(p)), 127.77 (s, Ph(m)), 130.51 (s, Ph(o)), 163.72 (s, Ph(i)), 191.56 (s, CO).
2.3.1.3 Data for [HgI2(Y)]2 (3)
White solid; yield: 89%, mp 180–182 °C. Anal. calcd. for Hg2I4O4S2C22H28(%): C, 19.88; H, 2.12; S, 4.82. Found: C, 19.81; H, 2.07; S, 4.59. IR (KBr disk): ν(cm−1)(assign.): 1629 (CO) and 824 (S+–C−). 1H NMR (DMSO-d6) δH ppm (multiciplicity of the signal, integr., assign.): 2.84 (s, 6H, S(CH3)2), 3.79 (s, 3H, OCH3), 5.24 (s, 1H, CH), 6.89 (d, 3JHH = 8.1 Hz, 2H, Ph), 7.79 (d, 3JHH = 8.1 Hz, 2H, Ph). 13C NMR (DMSO-d6, ppm) δC: 27.21 (s, S(CH3)2), 56.18 (s, OCH3), 65.64 (s, CH), 114.30 (s, Ph(p)), 127.86 (s, Ph(m)), 130.61 (s, Ph(o)), 163.83 (s, Ph(i)), 191.68 (s, CO).
3 Results and discussion
3.1 Synthesis
The-room temperature reactions of HgX2 (X = Cl, Br and I) with sulfonium ylide (prepared by reacting dimethyl sulfide with an acetone solution of 2-bromo-4′-methoxy acetophenone and treatment with aqueous NaOH solution) for 4 h (1:1 M ratio) in CH3OH gave complexes 1–3 (Scheme 2). Herein, we report comprehensive characterizations utilizing NMR spectroscopy and X-ray structural analysis. X-ray-quality crystals of complexes 1 and 2 were grown by the direct diffusion of methanol in the dimethyl sulfoxide solution over several days.
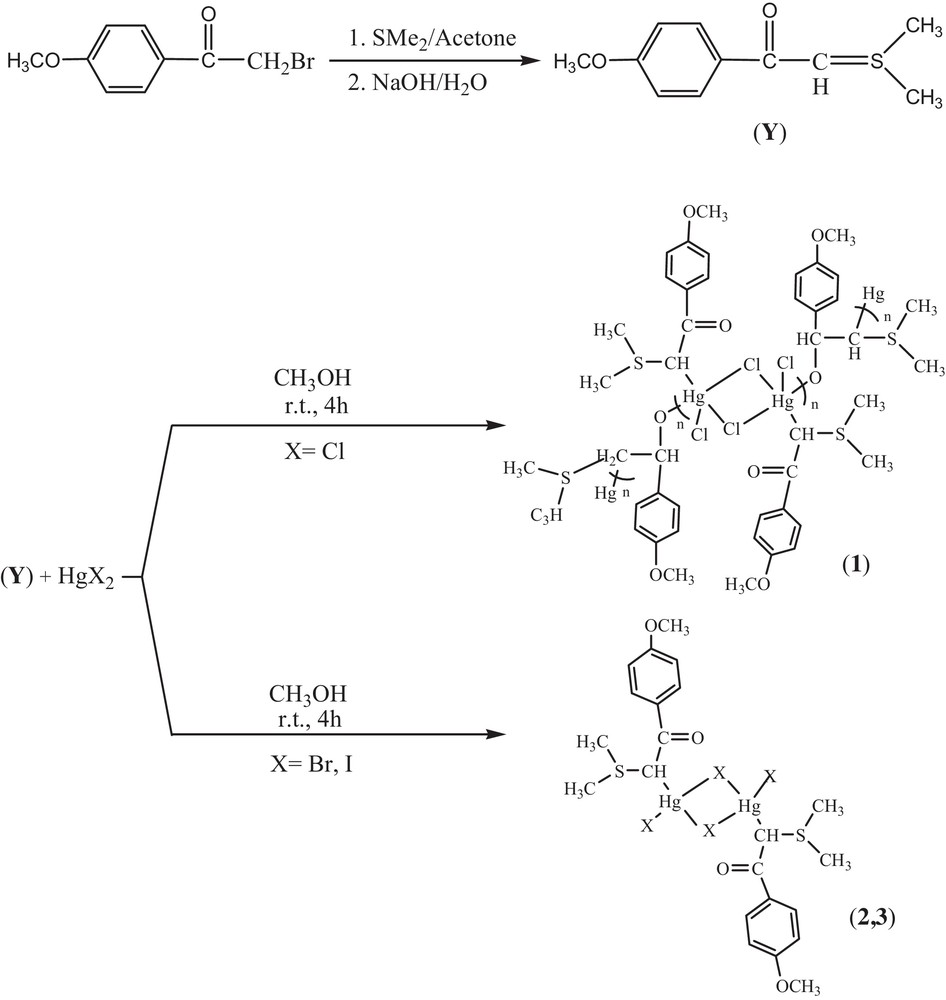
Synthetic route for the preparation of complexes 1–3.
3.2 Spectroscopy
In the infrared spectra, the carbonyl stretch, which is sensitive to complexation, occurs about 1583 cm−1 for the parent ylide, as in the case of other resonance-stabilized ylides [11]. The coordination of the ylide through carbon causes an increase in ν(CO), while for the O-coordination a decrease of ν(CO) is expected. The infrared absorption bands observed for all complexes at 1629–1649 cm−1 suggest a coordination of the ylide through the carbon atom. The ν(S+–C−) stretch, which is also evidenced for the coordination mode, occurs at 835 cm−1 in Me2S+–CH− and at 857 cm−1 in ylide. In the present study, the ν(S+–C−) values for all complexes were shifted to lower frequencies at 824–844 cm−1, suggesting a partial removal of electron density from the S–C bond due to coordination [11].
The 1H and 13C NMR signals for the SCH group of all complexes are shifted downfield compared to those of the free ylides, as a consequence of the inductive effect of the metal center. The appearance of single signals for the SCH group in 1H NMR at ambient temperature indicates that a fast equilibrium interconverting the two diastereoisomers is operating in solution. Since all spectra have been measured in strongly coordinating DMSO-d6, which competes with the ylide and can promote a fast bonding–unbonding of the ylide [8], an equilibration is occured between two diastereosiomers. In addition, no coupling to Hg (199Hg, 16.8% abundance, I = 1/2) was observed at room temperature in 1H and 13C NMR spectra. The 13C chemical shifts of the carbonyl carbon atoms in the complexes are around 191 ppm, compared to 182 ppm recorded for the same carbon in the parent ylide, indicating a decreased shielding of this carbon atom in the mercury complexes.
3.3 X-ray crystallography
The molecular structures of 1 and 2 were determined through single-crystal X-ray diffraction methods. Structural representations of complexes 1 and 2 are shown in Figs. 1 and 2. Crystallographic data and parameters concerning data collection and structure solution and refinement are summarized in Table 1 and selected bond distances and angles are presented in Tables 2 and 3, respectively.
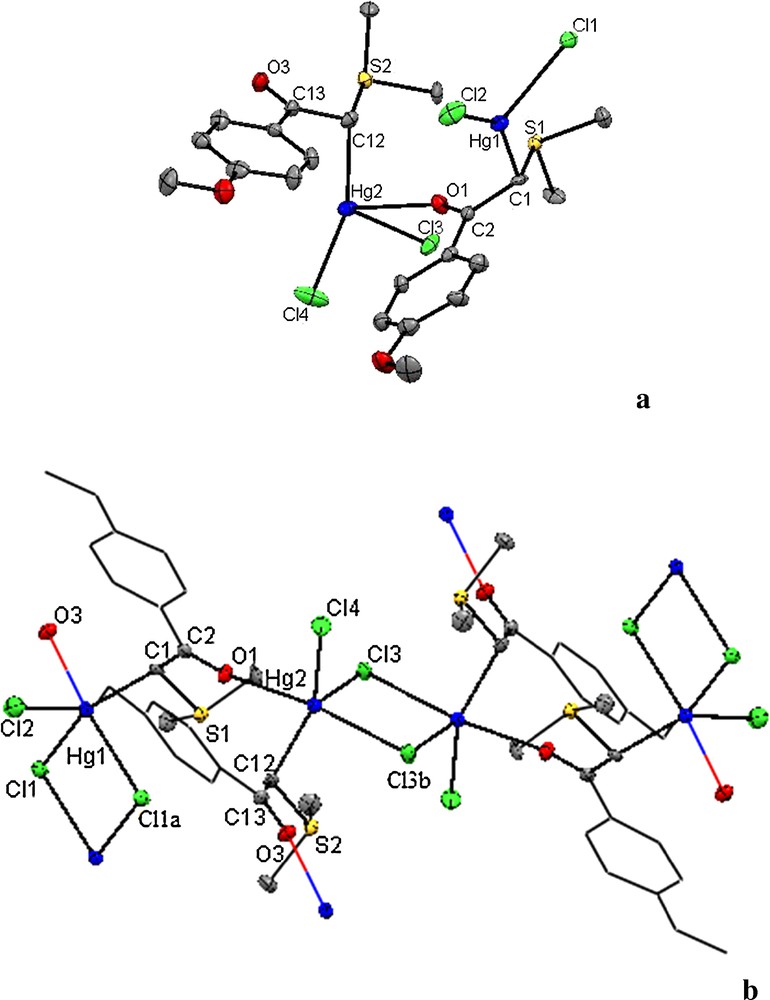
(Color online.) ORTEP view of the X-ray crystal structure of 1. (a) Asymmetric unit and (b) polymeric chain. H atoms have been omitted and the phenyl rings have been shown under a wire form for clarity. Symmetry code: a, –x, 1–y, 1–z; b: –1–x, 2–y, 1–z.
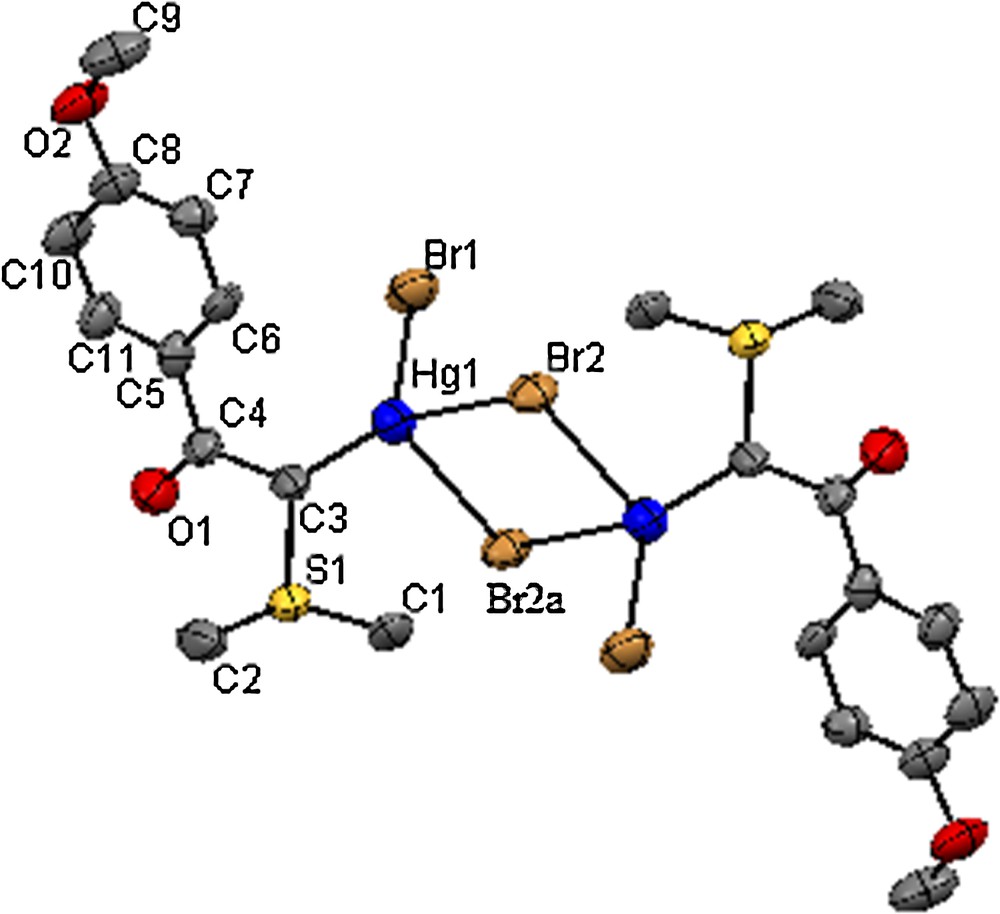
(Color online.) ORTEP view of the crystal structure of 2 as revealed by X-ray diffraction analysis. H atoms have been omitted for clarity. Symmetry code: –x,–y+1,–z+2.
Crystal data and refinement details for 1 and 2.
| CCDC | 881011 | 823067 |
| Formula | Hg2Cl4O4S2C22H28·2(C2H6OS) | Hg2Br4O4S2C22H28 |
| Formula weight | 1119.80 | 1141.36 |
| Temperature (K) | 130(2) | 293(2) |
| Wavelength (Ǻ) | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P21/c | |
| Unit cell dimensions | a = 10.1329(3) b = 12.4857(4) c = 15.3870(7) α = 110.370(4) β = 96.693(3) γ = 90.526(3) | a = 15.5940(7) b = 7.3471(4) c = 15.0885(7) – β = 115.877(3) – |
| Volume (Å3) | 1809.95(12) | 1555.37(13) |
| Z, Calculated density (mg/m3) | 2, 2.055 | 2, 2.437 |
| Absorption coefficient (mm−1) | 9.034 | 15.156 |
| F(000) | 1072 | 1048 |
| Crystal size (mm) | 0.27 × 0.08 × 0.06 | 0.40 × 0.30 × 0.20 |
| θ Range for data collection (°) | 2.73–30.01 | 2.70–26.00 |
| Limiting indices | –13 ≤ h ≤14 –17 ≤ k ≤16 –19 ≤ l ≤20 | –19 ≤ h ≤ 19 –8 ≤ k ≤ 9 –18 ≤ l ≤ 18 |
| Reflections collected | 9370 | 8130 |
| Unique | 7543 [R(int) = 0.0401] | 3046 [R(int) = 0.1154] |
| Completeness | 99.87% | 99.8% |
| Absorption correction | Gaussian | Numerical |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 9370/6/389 | 3046/0/155 |
| Goodness-of-fit on F2 | 1.057 | 1.075 |
| Final R indices [I > 2σ(I)] | R1 = 0.0436, wR2 = 0.1029 | R1 = 0.0796, wR2 = 0.2016 |
| R indices (all data) | R1 = 0.0601, wR2 = 0.1137 | R1 = 0.0903, wR2 = 0.2121 |
| Largest diff. peak and hole (eÅ3) | 5.86 and –1.66 | 2.758 and–6.020 |
Selected bond lengths (Å) for 1 and 2 and comparison of selected internuclear separations with triphenylphosphonium analogs.
| Bond lengths | 1 | [25] | Bond lengths | 2 | [25] |
| Hg1–C1 | 2.153(5) | 2.215(4) | Hg1–C3 | 2.158(12) | 2.218(11) |
| Hg1–Cl1 | 2.6417(15) | 2.418(14) | Hg1–Br2a | 2.9016(14) | 3.022(2) |
| Hg1–Cl1a | 2.8433(16) | 2.804(12) | Hg1–Br1 | 2.5031(16) | 2.559(2) |
| Hg1–Cl2 | 2.3795(15) | – | Hg1–Br2 | 2.7981(13) | 2.603(3) |
| Hg2–C12 | 2.166(6) | – | C3–C4 | 1.514(17) | – |
| Hg2–Cl3 | 2.6274(15) | 2.41(14) | C4–O1 | 1.199(15) | – |
| Hg2–Cl3b | 2.8579(15) | 2.884(12) | C3–S1 | 1.786(11) | – |
| Hg2–Cl4 | 2.3772(18) | 2.504(11) | Hg···Hg | 4.1131(8) | 3.994 |
| C1–S1 | 1.806(7) | – | |||
| C12–S2 | 1.796(7) | – | |||
| C2–O1 | 1.238(7) | – | |||
| C13–O3 | 1.223(7) | – | |||
| C1–C2 | 1.479(8) | – | |||
| C12–C13 | 1.465(8) | – | |||
| Hg···Hg | 3.8461(4) | 3.901 |
Selected bond angles (°) for 1 and 2.
| Bond angles | 1 | Bond angles | 2 |
| Cl1–Hg1–Cl1a | 91.03(4) | Br2–Hg1–Br2a | 87.64(4) |
| Cl1a–Hg1–Cl2 | 99.04(6) | Br2a–Hg1–Br1 | 103.34(5) |
| C1–Hg1–Cla | 93.90(17) | C3–Hg1–Br2a | 100.9(4) |
| C1–Hg1–Cl1 | 115.91(17) | C3–Hg1–Br2 | 112.0(3) |
| Cl2–Hg1–Cl1 | 96.97(6) | Br1–Hg1–Br2 | 101.08(5) |
| C1–Hg1–Cl2 | 144.39(17) | C3–Hg1–Br1 | 139.4(3) |
| S1–C1–Hg1 | 110.6(3) | S1–C3–Hg1 | 111.7(6) |
| O1–C2–C1 | 119.8(6) | O1–C4–C3 | 120.8(11) |
| Cl3–Hg2–Cl3b | 91.77(5) | ||
| Cl3b–Hg2–Cl4 | 96.50(6) | ||
| C12–Hg2–Cl3b | 94.28(16) | ||
| Cl4–Hg2–Cl3 | 97.07(7) | ||
| C12–Hg2–Cl4 | 144.29(17) | ||
| S2–C12–Hg2 | 110.5(3) | ||
| Hg1–O3–C13 | 160.2(4) | ||
| O3–C13–C12 | 120.7(6) |
There are two formula units in the asymmetric unit of this complex that are connected together through the oxygen of one of the ylides (see Fig. 1a), and two asymmetric units comprise the monomer, {HgCl2(Y)}4; these monomers connect together through halide bridges and make a two-dimensional polymer (see Fig. 1b). The X-ray analysis of complex 1 reveals a C-coordination of the ligand to the mercury ion. The structural analysis of the analog compound [HgCl2((Me)2SCHC(O)C6H4-p-Me)]2 [21] reveals a symmetric halide bridge (2.7223(16) and 2.7415(15) Å), while in complex 1, compared to [21], one of the bridging Hg–Cl bond lengths, 2.8433(16) Å, is considerably weakened due to the coordination of the ylidic oxygen to the metal ion. The other bridging Hg–Cl bond distance in 1, 2.6417(15) Å, is less than those found in [21], indicating a relatively strong Hg–Cl bond in this complex. As can be seen in Fig. 1, the ylidic oxygen and one of the bridging Cl atoms (Cl(3)–Hg2–O(1) 159.17(9)°) are located in the axial positions of the resulting distorted trigonal bipyramidal complex. In the equatorial positions, the Hg(II) atom is also surrounded by one ylidic carbon atom, two bridging and terminal Cl atoms with angles of 96.97(6)°, 115.9(2)° and 144.4(2)°. Thus, this compound can be considered as a pseudo five-coordinate complex. The Hg–O3 bond length, 2.948(4) Å, in complex 1 is significantly shorter than the sum of the van der Waals radii of Hg (1.70 Å) [23a] and O (1.54 Å) [23b] atoms and comparable with 2.927(4) A˚ [23] and 3.060 Å [24]. It is worth noting that in 1, the CO bond distance of 1.238(7) Å is longer than the experimental reference value of 1.223(8) and 1.231(8) Å in the analog compound [21].
The binuclear structure adopted by complex 2 is in contrast to the trinuclear structure exhibited by O-coordinated phosphorus ylide Ph3PCHC(O)Ph [25] to the mercury atom, but is similar to the structure of C-coordinated binuclear mercury(II) halide complexes of the phosphorus ylides Ph3PCHC(O)C6H4OCH3 (MBPPY) [26] and Ph3PCHC(O)OEt (EPPY)[27]. The Hg(II) center is four-coordinate with sp3 hybridization and the environment involves one short terminal Hg–Br bond, one Hg–C bond, and two asymmetric bridging Hg–Br bonds.
Although we could not obtain good qualified single crystals to determine the true structure of complex 3 by X-ray crystallography, nevertheless we suggest only a C-coordination to the ylide with a binuclear structure for complex 3, similar to that of 2, as a consequence of the symbiotic effects of the softer halogens. On the basis of DFT studies in the literature [24,28], the high HOMO energy indicates that the molecule can undergo an electrophilic attack with a large probability; hard molecules have a large HOMO–LUMO gap and soft molecules have a small one [28]. Thus, complexes including bromide and iodide as ligands are softer than the corresponding complex. This is completely consistent with the fact that a hard group makes a molecule hard and a soft group makes it soft.
It is generally accepted that the d orbitals of sulfur in a sulfonium group stabilize an adjacent carbanion to a greater extent than those of phosphorus in a phosphonium group [29,30]. However, in comparison with the analogous dinuclear mercury complexes with phosphorus ylides derived from triphenyl phosphonium (Table 2), the p–d overlap of the phosphonium group appears to be more pronounced than that with the dimethyl sulfonium groups incorporated into ylide Y. So, the dimethyl sulfonium ylides are more basic than the triphenyl phosphonium ylides, which is evident from the shorter Hg–C bond lengths, 2.153(5) Å in 1 and 2.158(12) Å in 2, in sulfonium ylide complexes compared with the equivalent distances in the phosphorus analogs (Table 2).
The terminal Hg–X (Cl4 and Br1) bond lengths, 2.3772(18) Å and 2.5031(16) Å, are less than those found in phosphorus analogs. The two bridging Hg–X (Cl1 and Br2) bonds, 2.6417(15) and 2.8433(16) Å in 1 and 2.7981(13) and 2.9016(14) Å in 2, are clearly asymmetric and much longer than in the phosphorus analogs.
The angles subtended by the ligands at the Hg(II) center vary from 87.64(4)° to 139.4(3)° in 2, indicating a much distorted tetrahedral coordination geometry. The widening of the Br–Hg–C angle from the tetrahedral angle must be due to the formation of a strong halide bridge between Hg atoms, which requires the internal Br–Hg–Br angles of 87.64(4)° to be considerably smaller. The two mercury atoms and two bridging halides in each case are perfectly coplanar. The internuclear distances between mercury atoms, 3.8461(4) Å in 1 and 4.1131(8) Å in 2, are close to those in phosphorus analogs, but this distance is much longer than the sum of van der Waals radii (3.0 Å) of the two mercury atoms [31], indicating the absence of significant bonding interactions between the mercury atoms in the molecular structures (see Table 2). The adaptation of binuclear structure in Hg(II) ylide complex 2 may be explained both by the preference of Hg(II) for coordination number four and the stability of the 18-electron configuration around Hg(II).
4 Conclusions
The present study describes the synthesis and characterization of some mercury (II) complexes of sulfonium ylides. On the basis of the physicochemical and spectroscopic data, it is clear that the sulfonium ylide ligands exhibit a monodentate C-coordination to the metal center. Nevertheless, the single-crystal X-ray analysis reveals an additional interaction including an O-coordination of the ylidic oxygen to the metal ion to form a pseudo five-coordinate complex for 1, while the use of bromo- and iodo-coligands affords four-coordinate binuclear complexes with only C-coordination. This difference can be accounted for by the greater electronegativity of chlorine versus its congeners, which enhances the Lewis acidity of the mercury centers to which it is bound, giving rise to a rare O-coordination, albeit with secondary Hg···O interactions. A comparison of important bond lengths with phosphium ylide analogs reveals a significant difference between the Hg–C bond lengths, which is attributed to greater Lewis basicity of dialkyl sulfonium ylide ligands versus triaryl phosphonium ylide ones.
Acknowledgements
We are grateful to the Bu-Ali Sina University, Hamedan, Iran for a grant and to Mr. Zebarjadian for recording the NMR spectra. F. Akhlaghi B. thanks the University of Melbourne, Australia for hosting her as an exchange visitor.