

1 Introduction
Cascade reaction implying different organocatalytic processes [1] have emerged as a useful and potent method for the efficient construction of important molecules, starting from easily available materials, in one pot [2]. As a matter of fact, numerous organocatalysts, either with cinchona alkaloid-[3], binol-phosphoric acid-[4], or thiourea-based [4a,5], are at present used with success in the asymmetric synthesis of a broad range of important complex organic compounds. However, while these organocatalysts may act as effective bifunctional asymmetric catalyst [6] and promote different types of reactions through different activation modes [7], they are still relatively little exploited in tandem or cascade process, involving asymmetric protonations as a stereo-defining step [8]. Asymmetric protonation of a transient prochiral species generated in situ from a synthetic operation, in the absence of metal components, are of great value because structurally complex optically active compounds can be obtained with high catalytic efficiency, stereoselectivity and atom economy. The most of these methods are based on protonation of a transient enol or enolate equivalents prepared through a nucleophilic addition to an α-substituted α,β-unsaturated carbonyl compound or to a ketene derivative, and surprising results have been obtained. In contrast, only a few examples based on process involving intramolecular rearrangement have been reported.
This account summarizes some important recent development in this area, and is organized on the basis of the synthetic strategies employed to generate the transient prochiral species: decarboxylation, Michael and ketene addition, N-heterocyclic carbene-catalyzed reactions and intramolecular rearrangement. In this respect, we highlight our recent contribution in this field of research. Also included in this account are the results of related investigations that have led to the development of novel methods based on tandem reactions initiated by an organocatalyzed enantioselective protonation, and on the application of asymmetric proton transfer catalysis (enantioselective [1,3]-proton shift) [6f].
2 Organocatalytic asymmetric protonation of transient enolates generated by decarboxylation
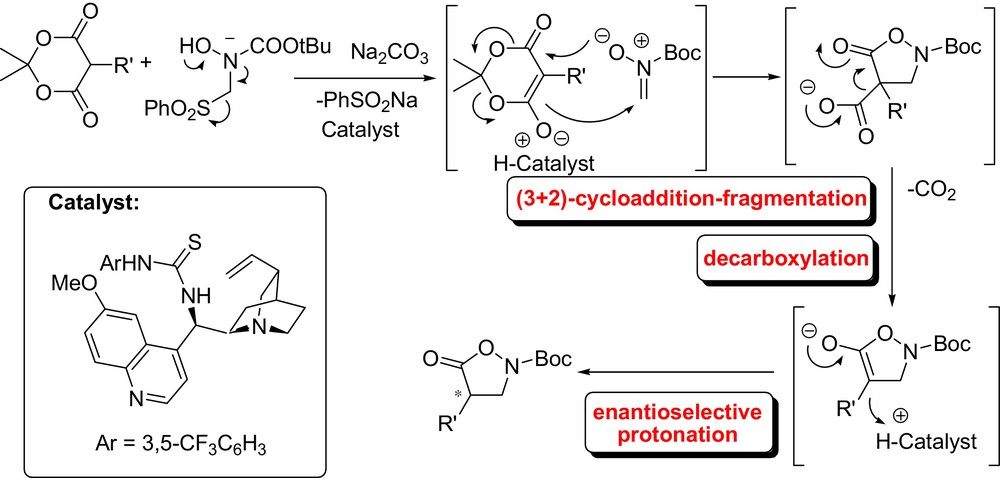
Organocatalytic enantioselective decarboxylation/reprotonation of malonate derivatives has been extensively investigated and the latest results have been covered in recent reviews [9]. Very recently, Brière et al. have designed an original enantioselective synthesis of isoxazolidones through an organocatalysed decarboxylative protonation process from readily available Meldrum's acid (Fig. 1). In this tandem reaction, the formal [3+2] cycloaddition-fragmentation with a N-Boc-nitrone is followed by an enantioselective protonation [10]. Up to 82% ee could be obtained with a cinchona-derived thiourea catalyst.
3 Organocatalytic asymmetric protonation of transient enolates (or enolates equivalents) generated by Michael addition
Seminal research on tandem sulfa-Michael addition-enantioselective protonation strategy, in the presence of a catalytic amount of a chiral amine, was reported by Pracejus et al. in 1977 [11]. In this respect, sometime thereafter Tan et al. reported a closely related chiral bicyclic guanidine-catalyzed addition of thiols and phosphine oxides to 2-phthalimidoacrylates [12].
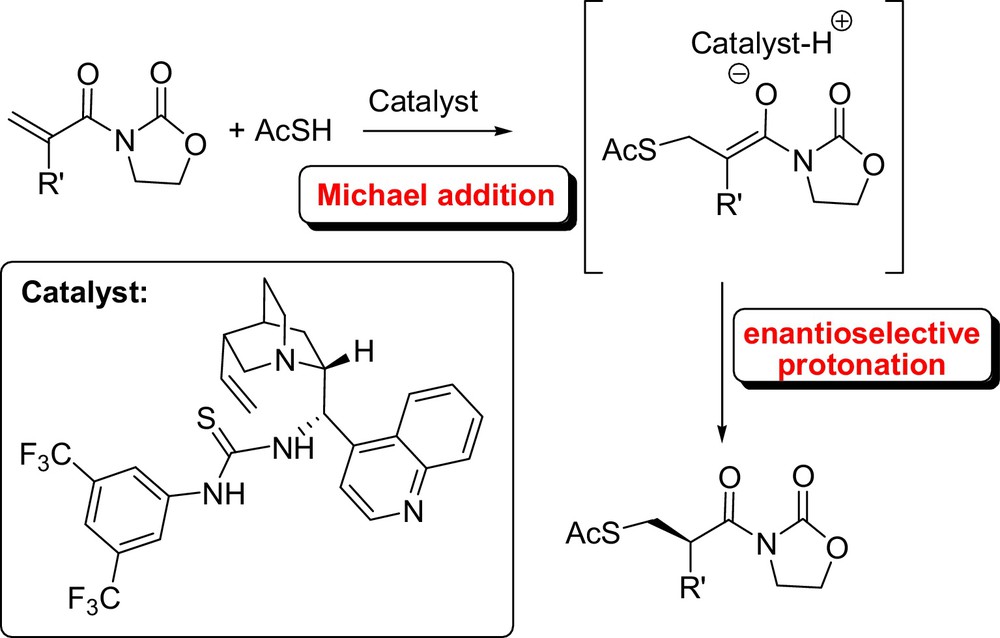
Singh et al. recently employed cinchona alkaloid derived thiourea catalysts in the enantioselective protonation of transient enolates generated by conjugate addition of thiophenol or thioacetic acid to a series of α-substituted N-acryloyloxazolidin-2-ones [13] (Fig. 2). Towards this end, Chen et al. screened various α-substituted acrylic derivatives and thiols in sulfa-Michael addition using chiral squaramides as hydrogen bonding organocatalysts. Among acrylic derivatives tested, α-benzyl acrylamide gave the best results yielding the corresponding products with excellent enantiomeric excess [14].
In the same context, Deng et al. employed bifunctional Cinchona alkaloid-derivatives as catalyst for the tandem asymmetric conjugate addition/protonation reactions of various cyclic and acyclic α-cyanoketone nucleophiles to 2-chloroacrylonitrile. In this reaction, two non-adjacent stereocenters could be generated with high stereocontrol [15]. Later, Xiao et al. published a highly enantioselective protocol for the synthesis of α-amino acids starting from 3-substituted oxindoles and ethyl 2-phthalimidoacrylate [16].
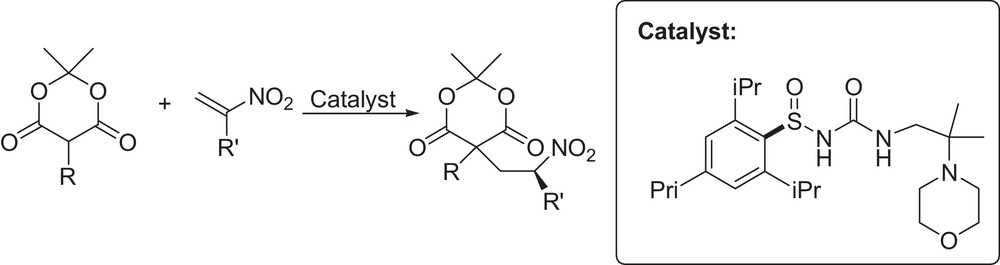
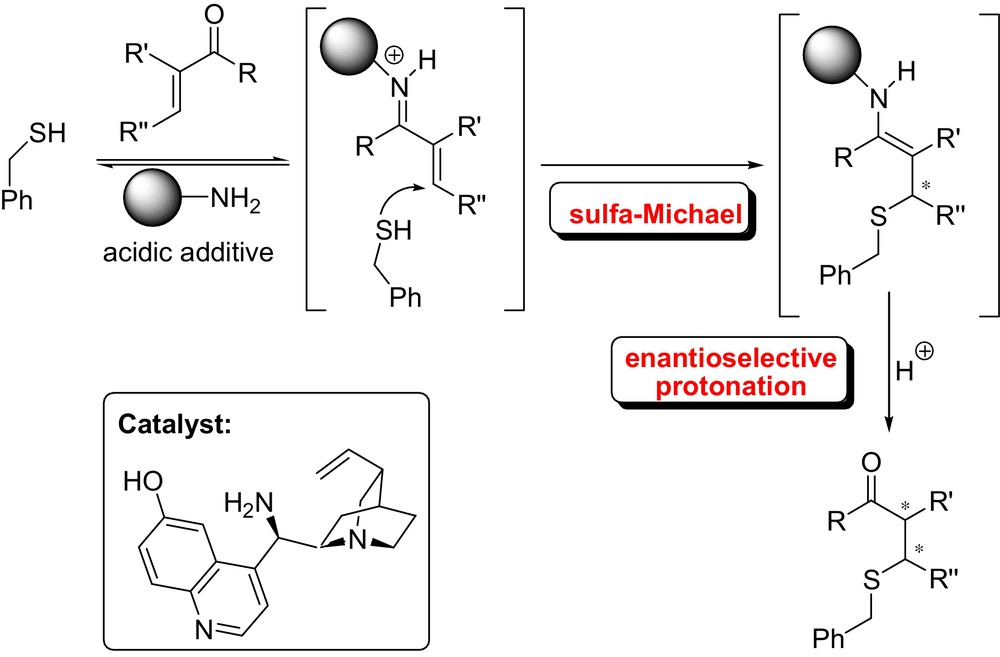
Ellman et al. successfully reported a highly enantioselective protonation of nitronates formed upon the addition of α-substituted Meldrum's acids to terminally unsubstituted nitroalkenes using a new type of N-sulfinyl urea catalyst. The resulting product was readily converted into the 3,5-disubstituted pyrrolidinones (Fig. 3) [17]. Similar chemistry has been investigated by Du and Yang who developed a squaramide-catalyzed asymmetric sulfa-Michael addition of thioacetic acid to α,β-disubstituted nitroalkenes[18]. Melchiorre et al. described a diastereodivergent asymmetric tandem sulfa-Michael additions/enantioselective protonation of alkyl thiols to α-branched enones using a cinchona-based primary amine [19] (Fig. 4). Careful optimization of reaction conditions and catalyst structure provided a convenient protocol for the synthesis of highly enantioenriched sulfa-Michael adducts. It is noteworthy that the choice of acidic additives and the reaction medium switches the sense of catalyst's stereoselection.
Luo et al. have developed the first (complementary to the enolate chemistry), enamine based, tandem Friedel–Crafts/enantioselective protonation, with a vicinal primary-tertiary diamine/toluenesulfonic acid catalyst, providing access to a range of important chiral indole derivatives [20a]. Interestingly, a mechanistic study by the authors have established that a water-bridged proton transfer and an unprecedented O–H/π interaction could be held responsible for the excellent stereochemical results obtained during the protonation step [20b]. The same protocol was also successfully applied in the conjugate addition of azoles to α-substituted vinyl ketones [20c]. Very recently, Zhang and Sun have reported an efficient intermolecular addition of nitroalkanes to activated enynes for the asymmetric synthesis of 2,3-allenoates catalyzed by cinchona-based thiourea catalyst. The reaction proceeded via an original tandem Michael addition/enantioselective protonation [21].
4 Organocatalytic asymmetric protonation of transient enolates generated by ketene addition
In pioneering work, Pracejus have studied the tandem nucleophilic additions on prochiral ketenes derivatives-enantioselective protonation, catalyzed by cinchona alkaloids [22] (Fig. 5). A similar reactions have been investigated by Simpkins et al., affording α-silyl thioesters with high degree of asymmetric induction starting from α-silyl ketenes [23]. Fu et al. have recently reported an enantioselective approach to optically active esters catalyzed by planar-chiral heterocycles involving the addition of alcohols to ketenes followed by an enantioselective protonation [24]. In an analogous manner, Fu developed also the method for the catalytic enantioselective addition of pyrroles to ketenes [25].

This process represented the first example of a planar-chiral heterocycle working as a chiral Brønsted acid catalyst. In the same area, Smith et al. have proposed a novel access to this type of products based on an asymmetric N-heterocyclic carbenes mediated addition of 2-phenylphenol to alkylaryl ketenes [26].
5 Asymmetric protonation of transient enolates and homoenolates generated by N-heterocyclic carbene catalysis
In the area of N-heterocyclic carbene catalysis, Rovis and de Alaniz reported an elegant highly enantio- and diastereoselective intramolecular Stetter reaction (Fig. 6). This approach involved the reaction of a trisubstituted Michael acceptors with either aromatic or aliphatic aldehydes by means of triazolinylidene carbene catalysts. This methodology involved the formation of a transient enolate, via intramolecular conjugate addition followed by diastereoselective proton transfer [27].

Glorius et al. have disclosed a simple and general NHC-catalyzed intermolecular enantioselective Stetter reaction with a highly stereoselective proton transfer as the key step that provides a series of amino acid derivatives (Fig. 7) [28].
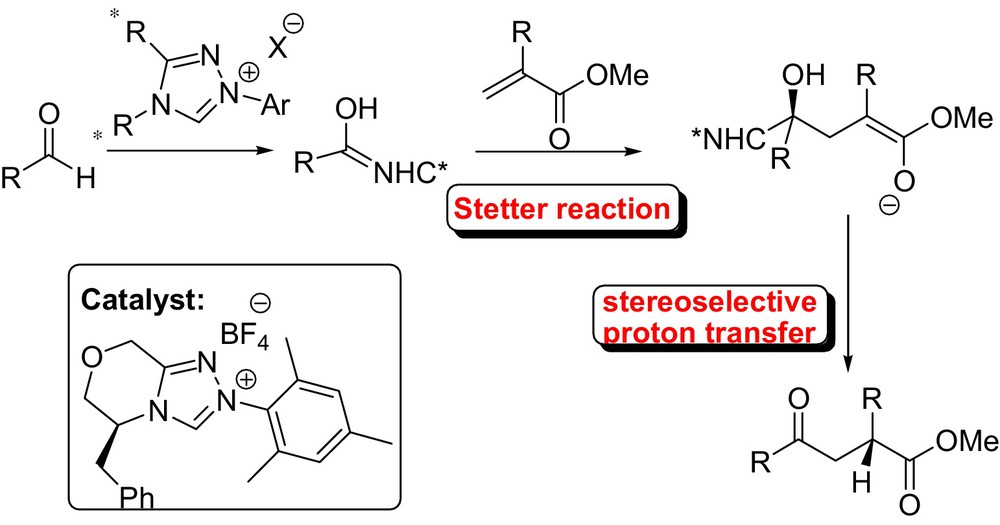
In the proposed reaction mechanism, the initial enantioselective attack of the Breslow intermediate on the Michael acceptor (methyl 2-acetamidoacrylate) was followed by a highly stereoselective intramolecular proton transfer that relays the stereochemical information previously generated. Recently, the same research group reported also a highly enantioselective protocol for the reaction of simple α,β-unsaturated esters (acrylates) without a second activating group [29].
Anyway, an early example of an intramolecular stereoselective protonation of aldehyde-derived enolates via N-heterocyclic carbene catalysis was reported by Kalesse et al. in 2010, in the course of their synthetic endeavors towards the natural product angiolam [30]. Rovis et al. have developed the synthesis of α-haloesters based on an enantioselective protonation of a transient NHC-generated azolium enolates (Fig. 8).
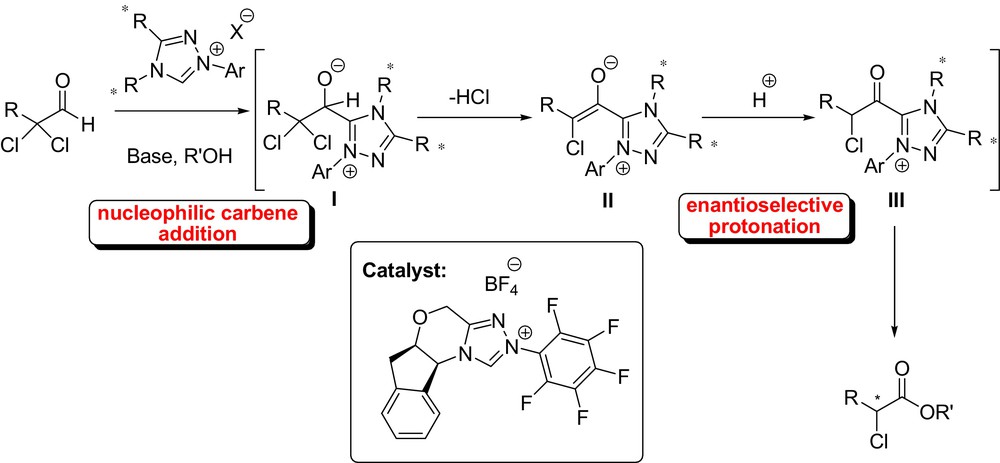
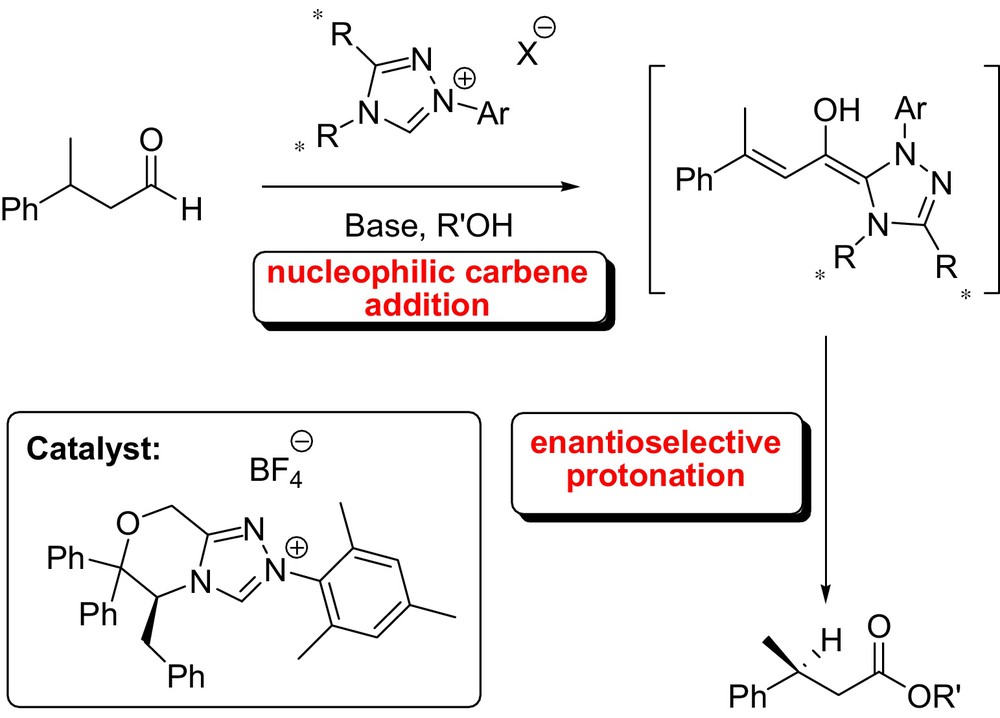
The tandem reaction, catalyzed by a chiral triazolinylidenecarbenes in the presence of phenol, afforded the species I from an α,α-dichloro aldehydes, which subsequently underwent an elimination of HCl providing the corresponding azolium enolate II. Protonation of this intermediate and subsequent esterification provided α-haloesters in excellent ee and high yields. The process had a wide scope affording a range of α-haloesters with various substituents [31]. In the same context, Scheidt et al. described an interesting application of carbene catalysis in enantioselective protonation of homoenolate equivalents (Fig. 9). Although the enantioselectivity was not high, this work is an important demonstration that enantioselective protonation can occur at a site remote from the chiral catalyst [32].
6 Organocatalytic asymmetric protonation of transient enolates generated by intramolecular rearrangement
In 2007, Rueping et al. developed the first Brønsted acid-catalyzed asymmetric tandem cyclization–protonation reaction (Nazarov cyclization), which provides a number of different cyclopentenones in good yields and with high enantioselectivities (67–78% ee) (Fig. 10). The proposed mechanism of this transformation involves the formation of a transient enolate by an 4π electrocyclization followed by a chiral phosphoric acid diester-promoted kinetic protonation.

In addition, it should be noted that this process employed a low catalyst loading of only 5 mol% [33a]. In a later report, the same research group showed that acyclic ether derivatives could also be used in organocatalyzed Nazarov cyclizations. This finding resulted in an operationally attractive method for the synthesis of simple cyclopentenone derivatives without any fused tetrahydropyrane ring [33b].
In 2009, Bolm et al. developed the enantioselective protonation of a transient enediol prepared by an intramolecular Cannizzaro-type rearrangement of hydracted arylglyoxal yielding optically active mandelic acid methyl ester derivatives with up to 83% ee (Fig. 11). This novel process was catalyzed by a cinchona alkaloid dimer in combination with an achiral thiol. The catalyst could be easily recovered and reused without significant loss of chemical yield or enantioselectivity [34].

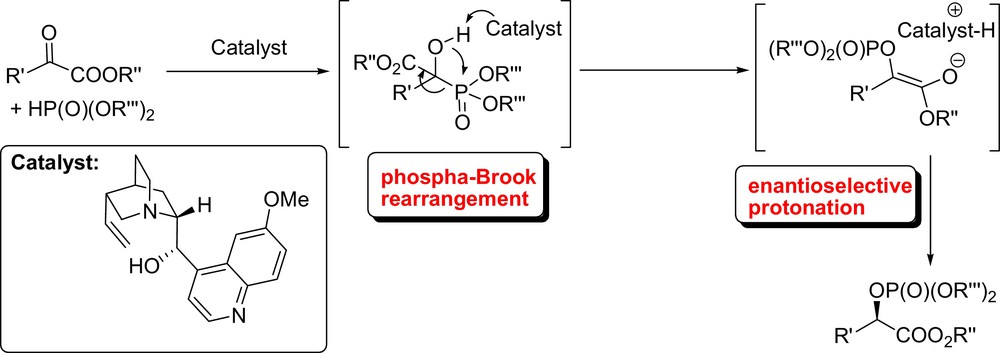
Nakamura and Hayashi reported a highly enantioselective protonation of ester enolates prepared via phospha-Brook rearrangement (Fig. 12). In this work, 3.0 equivalent of diphenyl phosphite reacted with an ethyl phenylglyoxalate in the presence of cinchona alkaloids catalyst and a stoichiometric amounts of Na2CO3 to afford a transient α-phosphonyloxy enolate followed by an enantioselective protonation. A series of optically active phosphoric esters having secondary alcohols were achieved with good yield and excellent enantioselectivities using commercially available cinchona alkaloids [35].
In this context, we investigated the enantioselective organocatalytic rearrangement of α-acyloxy-β-ketosulfides to α-acyloxy thioesters derivatives which involves the generation of a transient enolate though a proton abstraction from a terminal carbon by means of cinchona alkaloids, followed by an in situ enantioselective protonation (Fig. 13). Remarkably, high levels of yields and good to high enantioselectivities were observed for the products arising from the reaction of a range of α-acyloxy-β-ketosulfides. It is noteworthy that the Pummerer reaction of β-ketosulfoxides, followed by acyl migration is one of the most useful methods for the preparation of α-acyloxy thioesters which can be easily transformed into sulfur-free biologically active products, such as α-hydroxy acids, amides, esters without racemization [36].
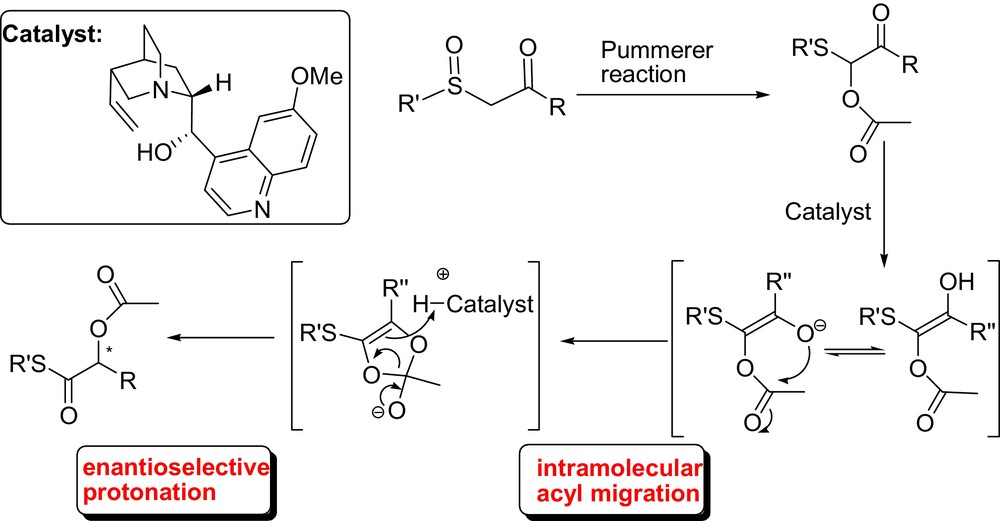
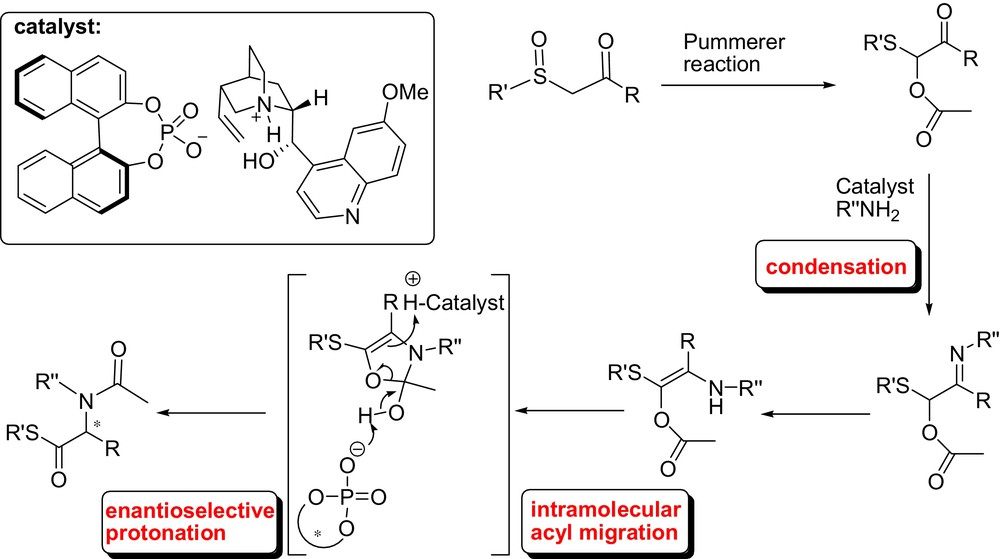
Then, as a logical extension of our work, we have also demonstrated that the same concept could be applied to the preparation of the corresponding chiral α-amino thioester derivatives in good yield and with moderate enantioselectivities.
The method was based on an unprecedented and conceptually novel chiral Brønsted base/Brønsted acid-catalyzed tandem condensation-intramolecular rearrangement-protonation. Although the degree of enantioselectivity observed for this reaction was moderate, these preliminary results formed the basis for further developments (Fig. 14) [37]. As a matter of fact, in the same context, very recently we have reported the first attempts towards a catalytic and enantioselective Amadori–Heyns-type rearrangement and its application for the synthesis of optically active α-amino ketones. The Amadori–Heyns rearrangements, better known in carbohydrate and food chemistry, allows for simple and selective introduction of an amine onto C-1 of a α-hydroxy carbonyl moiety and does not require any protecting group manipulation [38]. A mechanism involving a tandem condensation-intramolecular rearrangement-proton transfer reaction catalyzed by cinchona alkaloids has been proposed. The products have been isolated in good to high yields and up to 81% ee (Fig. 15) [39].

The methodology reported complements among the others, the alternatives recently reported based on the enantioselective “electrophilic α-amination” of carbonyl compounds [40]; and result as another example of the more appreciate potential of cinchona scaffolds to induce stereocontrol in organocatalytic reactions. Most notably, the scope of the reaction could be extended to the first catalytic enantioselective synthesis of α-amino cyclobutanones from the readily available α-hydroxy cyclobutanone and N-alkyl-anilines [41a]. Moderate to high enantioselectivities (up to 81% ee) were obtained with a series of N-methylanilines with different substitution patterns (Fig. 16). In this case, the reaction could be rationalized by assuming the mechanism shown in Fig. 17. The biscinchona alkaloid catalyzes the generation of an 1,2-enaminol from the α-hydroxy cyclobutanone and an N-alkyl-aniline followed by an in situ enantioselective enol-keto tautomerisation. Water-mediated proton transfer provides the product and releases the catalyst back into the cycle. In continuation of our work, we have also found that the methodology could be applied successfully to the stereoselective synthesis of cyclobutanone α-amino acid ester derivatives [41b].
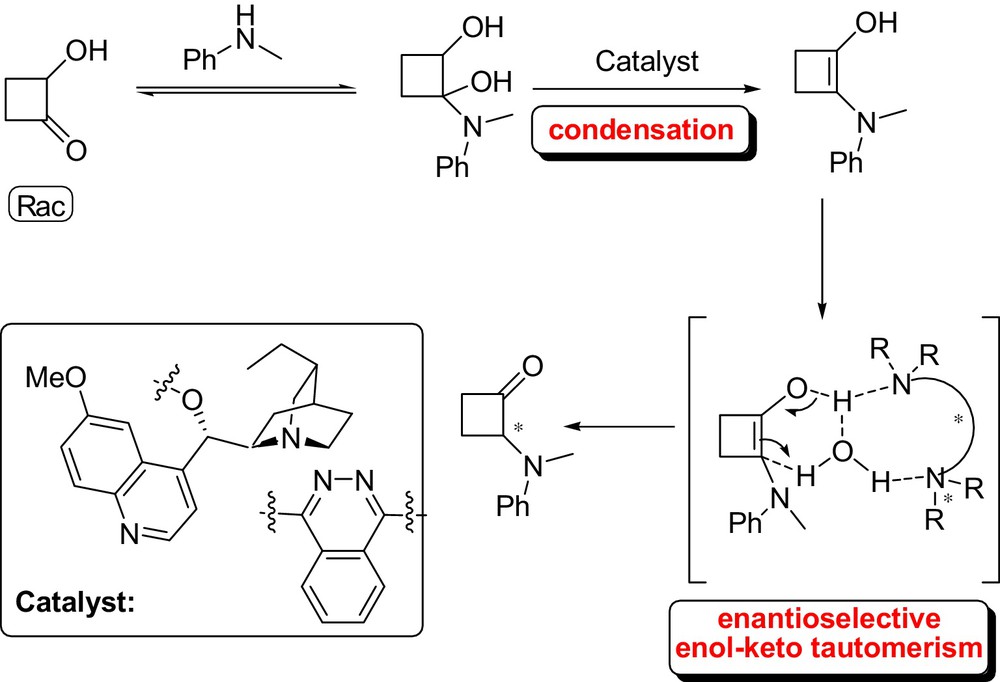
It is noteworthy that simple optically active α-amino cyclobutanones are more difficult to access by direct electrophilic α-amination [42]. Furthermore, synthetic methods which rely on the use of cyclobutane based structural moiety are of considerable value, because these compounds are key scaffolds of a large number of natural products and versatile building blocks in organic synthesis owing to their inherent ring strain [43].
7 Tandem reactions initiated by an organocatalyzed enantioselective protonation
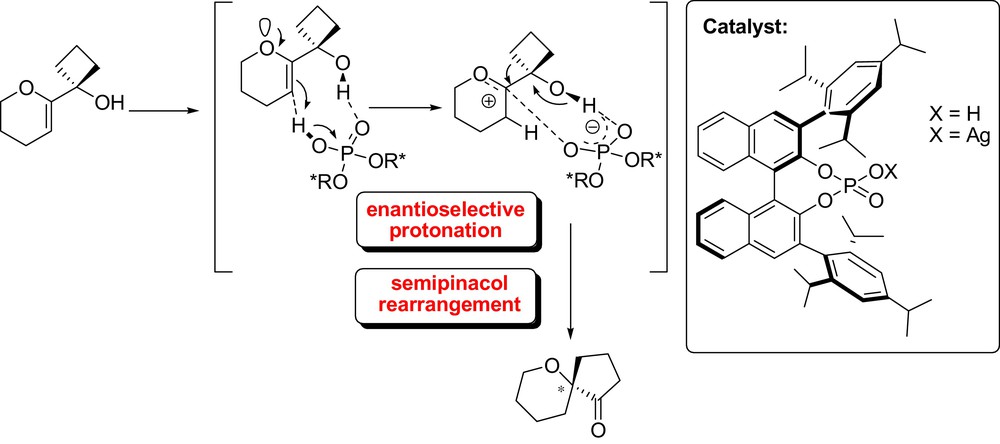
In the context of tandem reactions initiated by an organocatalyzed enantioselective protonation, Tu et al. have reported an elegant chiral phosphoric acid-catalyzed enantioselective tandem protonation-semipinacol rearrangement (1,2-carbon migration involving hydrogen bonding) of 2-oxo allylic alcohols for the synthesis of chiral spiroethers bearing an oxo-quaternary carbon stereocenter in high yields and excellent enantioselectivities (Fig. 17) [44]. On the other hand, Lee and List developed an unprecedented deracemization via a tandem catalytic asymmetric protonation induced-cyclization of ketene dithioacetals using commercially available chiral phosphoric acid catalyst (Fig. 18). Racemic α-aryl hydrocoumarin derivatives, easily obtained by condensation of salicylaldehyde derivatives and α–aryl acetyl chloride followed by reduction were shown to be viable starting materials for this transformation, yielding thioacetal-protected high enantioenriched hydrocoumarin [45].
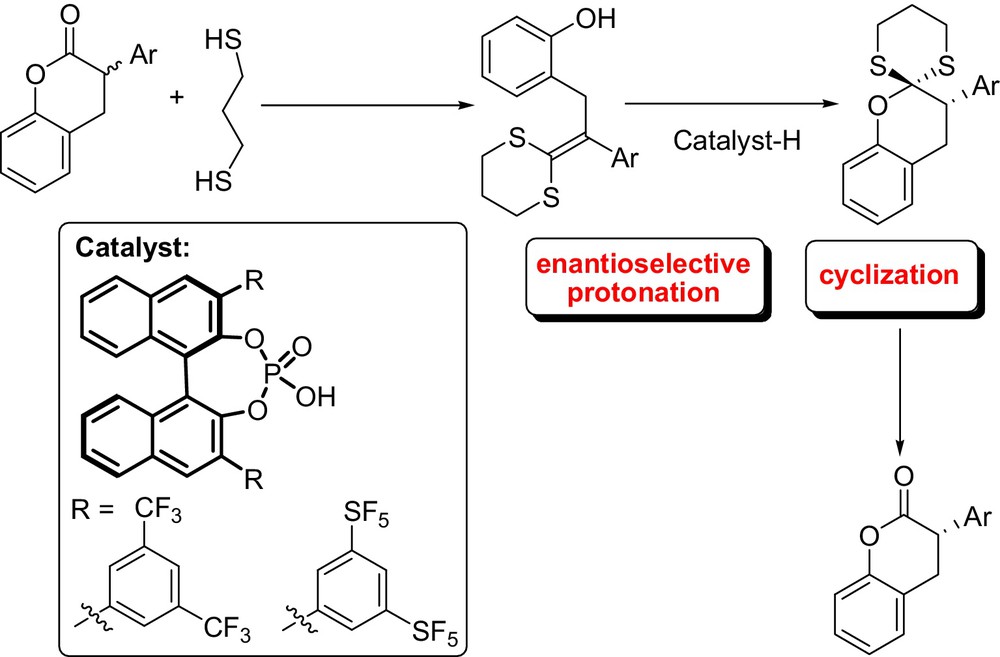
Hydrocoumarin and related chroman derivatives are present in a myriad of natural products with important antioxidant and other biological activities. Notably, the reaction was employed for accessing the optically active non-steroidal estrogen (S)-Equol. The same research group also studied the enantioselective protonation of silyl ketene imines catalyzed by either STRIP or TRIP via dynamic kinetic resolution. Moreover, this approach was applied to the synthesis of highly enantioenriched α-branched nitriles [46].
8 Organocatalyzed tandem process involving an enantioselective proton transfer ([1,3]-proton shift)
Recently Shi et al. have studied a general organocatalyzed tandem process involving an enantioselective proton transfer for the enantioselective synthesis of a range of α-amino acid derivatives (Fig. 19). The authors have shown a new synthetic method that mimic enzyme-catalyzed transamination via a catalytic enantioselective ketamine tautomerization to the corresponding aldimine. High enantiomeric excess were obtained for a variety of α-keto esters using a quinine-derived catalyst [47].
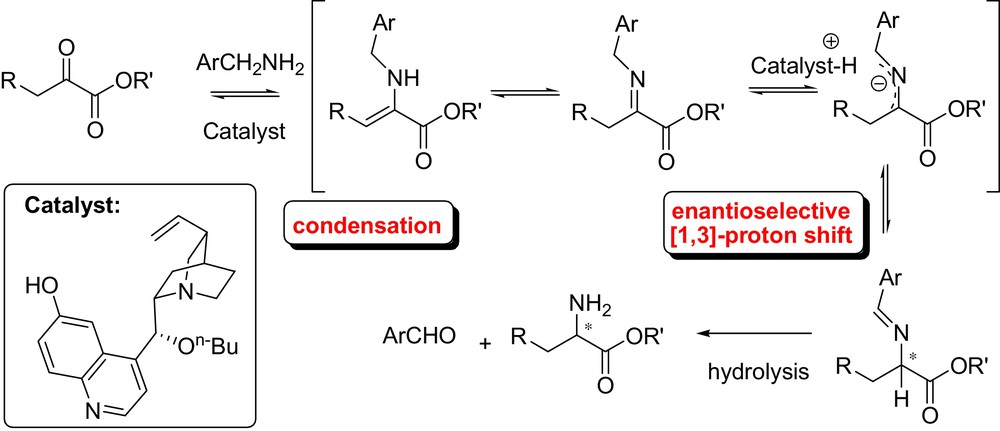
However, an early example of asymmetric [1,3]-proton shift mediated by cinchona alkaloids was first reported by Soloshonok et al. in 1997 (Fig. 20) [48]. The authors developed a moderately enantioselective proton transfer for the preparation of optically active fluorinated β-amino acids.
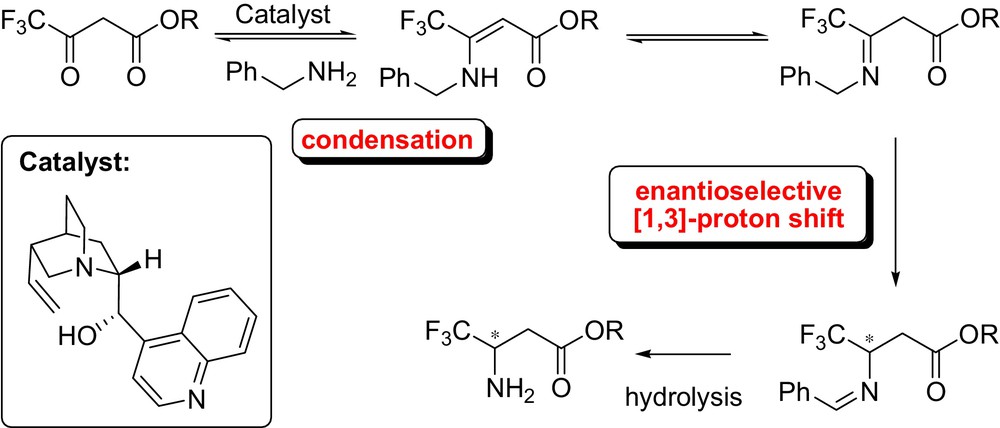
This biomimetic synthesis of β-fluoroalkyl-β-amino acids from β-keto esters consisted in the thermodynamic equilibration of N-benzyl enamines into N-benzylidene derivatives, isomerized into the corresponding aldimines, and then hydrolyzed to afford the free amino group. Following this study Plaquevent et al. further explored the reaction achieving good enantiomeric excesses. In addition, a comprehensive mechanistic study demonstrated that the deprotonation step was both rate and asymmetric determining step [49].
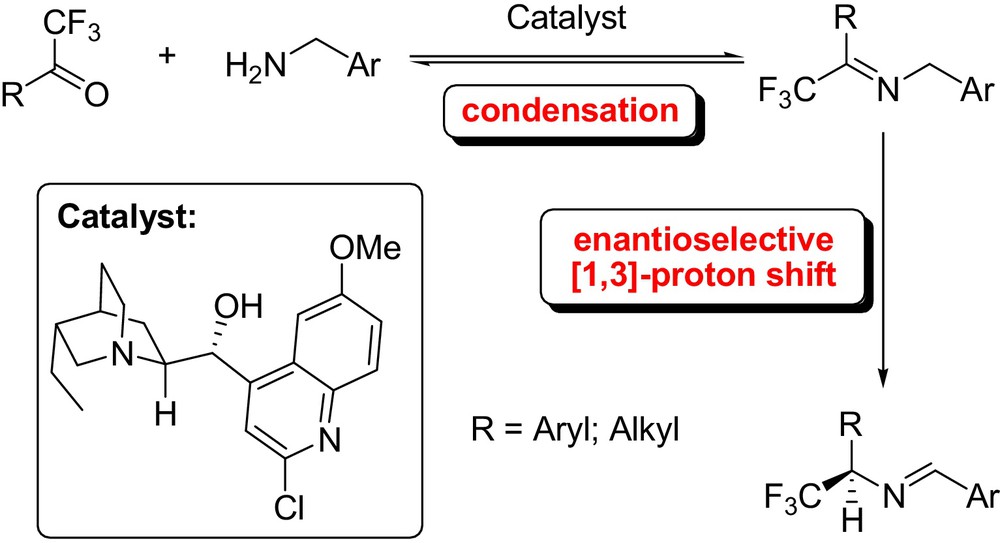
Soloshonok described also the successful catalytic asymmetric synthesis of α-(trifluoromethyl)benzylamine via cinchonidine derived base-catalyzed [1,3]-proton shift reaction [50]. Very recently, Shi [51] and Deng [52] independently used a conceptually similar approach for the highly enantioselective chiral organic base-catalyzed [1,3]-proton shift of trifluoromethyl imines. It is noteworthy that alkyl trifluoromethylated imines also proved to be viable substrates in these protocols (Fig. 21). Interestingly, Shi et al. applied the above-mentioned procedure to generate optically active amines from inactivated ketones [53].
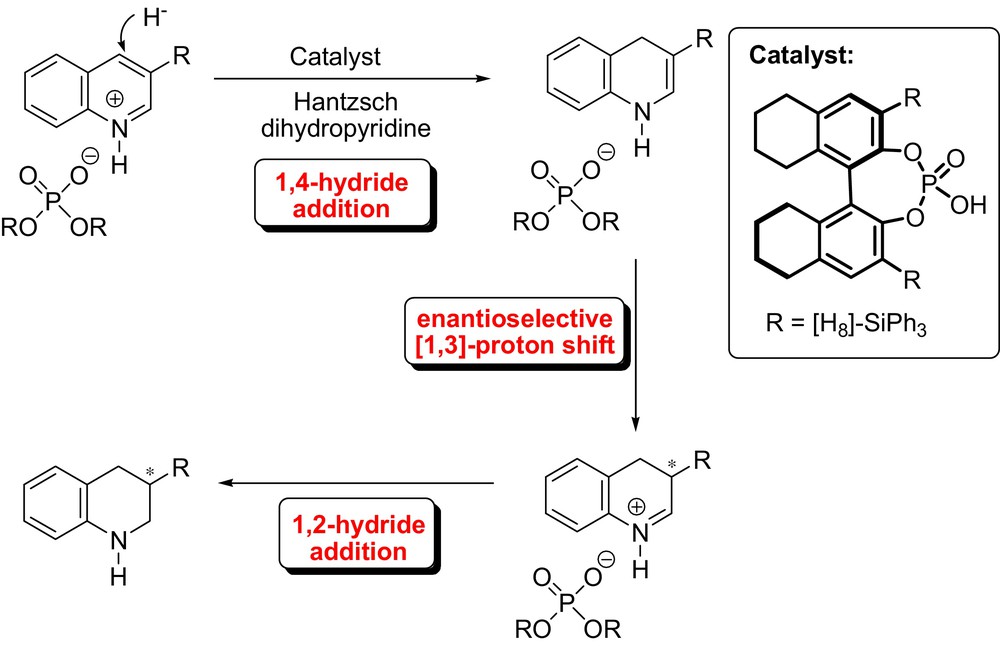
In another context, Rueping et al. developed a catalytic enantioselective enamine protonation in the course of quinolone reductions (Fig. 22). In this organocatalytic transfer hydrogenation, the activation of the quinolines is achieved by a catalytic protonation via a chiral Brønsted acid, which subsequently allows a cascade hydrogenation involving an enantioselective tandem [1,3]-proton shift-1,2 hydride addition initiated by a 1,4-hydride addition. A variety of tetrahydroquinolines or octahydroacridines were obtained in good yields and with high enantioselectivities [54].
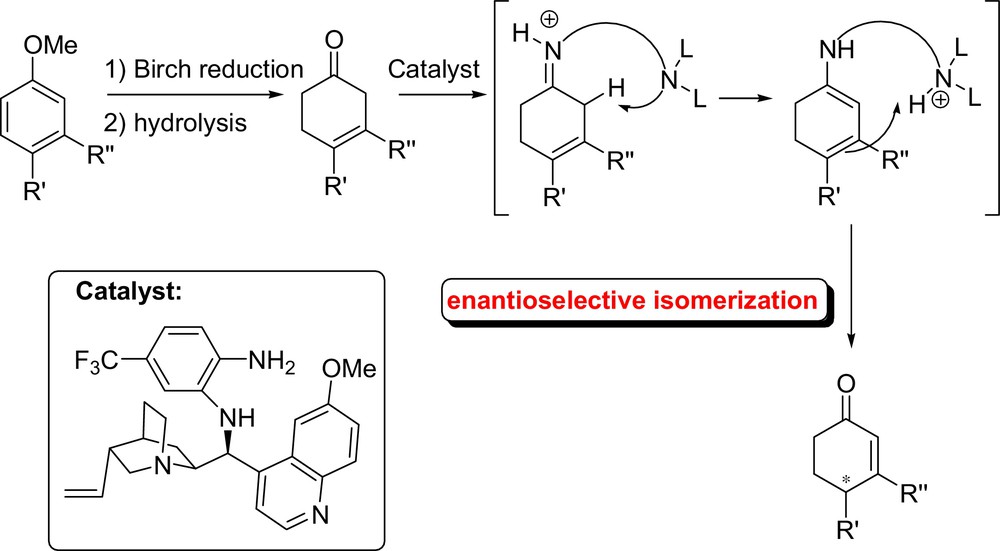
Also of interest in this field is the asymmetric approach toward cyclohex-2-enones from anisoles via an unprecedented enamine catalyzed enantioselective isomerization developed by Li Deng (Fig. 23). This methodology has been extended to the total synthesis of the natural product (–)-isoacanthodoral [55]. The same research group studied also the enantioselective olefin isomerization of β,γ-unsaturated butenolides into the corresponding α,β-unsaturated butenolides, catalyzed by cinchona alkaloids [56].
9 Conclusions
In the past few years, significant advances in the development of efficient organocatalyzed asymmetric protonation protocols have been made. Among them, recently tandem or cascade process, where the protonation step is the stereo-defining step, has received some attention. Regarding the accessibility of the starting materials, the simplicity of the operation, and high yields and stereoselectivities, such synthetic approach have proven to be a useful tool that allows the preparation of a wide range of optically active building blocks. Although these catalytic methodologies offer many advantages, some mechanistic aspects have still not received an entirely satisfactory explanation and in some cases, more effort might be necessary to achieve better enantioselectivity and to expand the substrate scope. Therefore, future studies should focus on the further elucidation of the mechanistic details of protonation of the various transient prochiral species. Because new concepts for enantioselective protonations continue to be disclosed, it is likely that increasingly ambitious applications of this chemistry will be described in the literature.
Acknowledgments
Financial support from the MIUR, Rome, and by the University of Cagliari (National Project “Stereoselezione in Sintesi Organica Metodologie ed Applicazioni”) and from CINMPIS and FIRB 2008 is gratefully acknowledged.